
Open Access, Volume 11
Diagnostic mimics of ocular myasthenia gravis: A 20-year case of misdiagnosis
Nithisha Thatikonda, MD*; Princy Lukhi, MD; Komal Hafeez, MD
Department of Neurology, University of Texas Medical Branch at Galveston, USA.
Nithisha Thatikonda
Department of Neurology, University of Texas Medical Branch at Galveston, PGY-4, Neurology Resident 301, University Blvd, Galveston, TX, USA.
Email: nithatik@utmb.edu
Received : September 19, 2025,
Accepted : October 15, 2025
Published : October 31, 2025,
Archived : www.jclinmedcasereports.com
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Thatikonda N (2025)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Thatikonda N, Lukhi P, Hafeez K. Diagnostic mimics of ocular myasthenia gravis: A 20-year case of misdiagnosis. Open J Clin Med Case Rep. 2025; 2387.
Introduction
Diagnosing rare conditions can often be clouded by atypical symptoms that mislead clinicians, delaying accurate identification and treatment. Ocular Myasthenia Gravis (OMG) exemplifies this diagnostic challenge, presenting with hallmark symptoms such as ptosis, diplopia, and ophthalmoplegia—features shared with other neuromuscular disorders [1]. Typically, OMG is diagnosed through a combination of clinical presentation, positive autoantibody testing, and neurophysiological evidence [2]. However, a subset of patients lacks detectable autoantibodies, a condition known as seronegative OMG, which complicates diagnosis, especially in the absence of supportive neurophysiological findings [3].
In such cases, the reliance on clinical judgment alone increases the risk of misdiagnosis or delayed diagnosis, with studies reporting error rates as high as 13% [4]. These misdiagnosed patients often experience a chronic, static, and treatment-refractory course that contrasts with the characteristic fluctuating symptoms of true OMG [4]. Misdiagnosis not only prolongs patient suffering but also subjects them to unnecessary, potentially harmful interventions [4].
Herein, we present the case of a patient who was misdiagnosed with OMG and whose diagnosis was re-addressed after 20 years of seeking medical care. Additionally, we discuss the diagnostic mimics of OMG that should be considered as differential diagnoses, especially in patients with chronic symptoms of ptosis and ophthalmoplegia and a treatment-resistant disease course.
Case Presentation
A 71-year-old man presented with a 20-year history of bilateral eyelid closure, more pronounced on the right (R>L), and restricted eye mobility. In 2009, he visited an ophthalmology hospital, where fundoscopy and optical coherence tomography revealed no abnormalities. Subsequently, he was diagnosed with ocular myasthenia gravis by her primary care physician and prescribed oral pyridostigmine (60 mg Q8H) and prednisone (30 mg QD). However, his symptoms showed no improvement. Due to financial constraints, he did not pursue further medical evaluation and continued taking the medications at the same dosage. In 2022, he discontinued prednisone, perceiving no significant improvement in his symptoms while on the medication.
In 2024, he sought medical attention at our hospital due to worsening ptosis, which caused vision problems and recurrent falls. At the time of his admission in March 2024, neurological examination revealed complete ptosis of the right eyelid and incomplete ptosis of the left eyelid. There was extraocular movement restriction in all directions in both eyes, except for mildly preserved abduction in the right eye. Bilateral upper and lower extremity muscle strength was intact, graded as 5/5. Serological tests for myasthenia gravis, including anti-AChR (acetylcholine receptor), anti-MuSK (Muscle-Specific Kinase), anti-Titin, and anti-LRP4 (low density lipoprotein receptor-related protein 4) antibodies, were negative. Laboratory investigations revealed an elevated serum lactate level of 19.0 mg/dL (normal range: 4.2-17.0 mg/dL) and a lactate/pyruvate ratio of 21 (normal range: <10). Levels of serum Lactate Dehydrogenase (LDH), Creatine Kinase (CK), and Creatine Kinase Isoenzyme (CK-MB) were within normal ranges. Other routine blood and biochemical tests were unremarkable. Electromyography (EMG) revealed no abnormalities, with negative findings on the repetitive electrical stimulation test. Further investigation with mitochondrial genome sequencing revealed homozygous mutations in the Ribonuclease H1 (RNASEH1) gene. The patient declined a muscle biopsy for confirmatory testing. Based on his clinical presentation and genetic testing results, he was diagnosed with RNASEH1-related progressive external ophthalmoplegia [5].
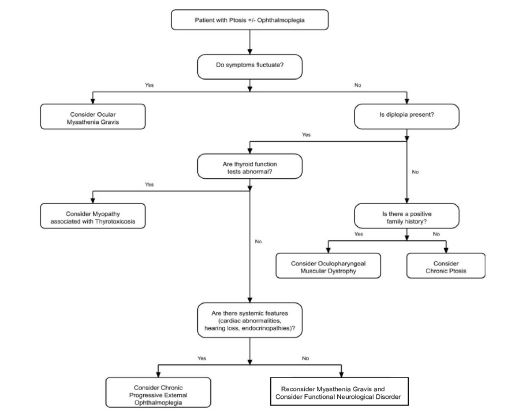
Figure 1: Diagnostic algorithm to approach a patient presenting with ptosis with or without ophthalmoplegia.
Table 1: Ocular myasthenia gravis diagnostic mimics with typical features and clues for differential diagnosis. Overlapping symptoms with OMG (ptosis, ophthalmoplegia) are shown along with distinguishing features and diagnostic clues. See text for details and references.
| OMG mimic | Etiology | Overlapping features with OMG | Distinguishing features | Clue for diagnosis |
|---|---|---|---|---|
| CPEO (Chronic Progressive External Ophthalmoplegia) | mitochondrial DNA mutations | ophthalmoplegia and ptosis |
|
|
| OPMD (Oculopharyngeal Muscular Dystrophy) | autosomal dominant (GCG expansion in the PABPN1 gene) | ptosis (usually trinucleotide repeat related), and variable ophthalmoplegia |
|
|
| Myopathy associated with thyrotoxicosis | catabolic effects of hyperthyroidism | ptosis and diplopia |
|
|
| Chronic ptosis | multiple causes, including myopathic and neuropathic and mechanical causes | ptosis |
|
|
| Functional Neurological Disorder (FND) | psychogenic or functional origin | variable ptosis and possible diplopia |
|
|
Discussion
OMG is an autoimmune disorder of the neuromuscular junction that causes fluctuating eyelid drooping (ptosis) and double vision (diplopia). Because many other conditions can produce similar eye findings, it is important to recognize “myasthenic mimics” – disorders with ptosis and ophthalmoplegia that overlap with OMG but have different causes and features. (Table 1) summarizes common OMG mimics, their etiologies, and key clues that distinguish them from true ocular myasthenia. In general, mimics tend to have non-fluctuating, progressively worsening symptoms and no response to myasthenia-specific tests (e.g. no decrement on repetitive nerve stimulation and no improvement with edrophonium). Systemic features, genetic testing, and imaging often provide diagnostic clues. We discuss each category of mimic in detail, emphasizing overlapping and distinguishing features and how to approach diagnosis.
Chronic Progressive External Ophthalmoplegia (CPEO)
CPEO is a rare mitochondrial myopathy characterized by insidious, bilateral, and symmetrical ptosis with slowly progressive ophthalmoparesis [6,7]. Prevalence is very low (approximately 1-2 per 100,000) [1]. CPEO can present in various forms, primarily classified based on underlying genetic mutations and clinical features. The most common form, known as classic CPEO, is caused by mutations in mitochondrial DNA (mtDNA) [6]. The condition may occur sporadically or be inherited, indicating both sporadic and familial forms of the disease [7]. While CPEO typically manifests in young adults between their 20s and 40s, it can also present later in life [7]. Unlike OMG, CPEO symptoms are not fluctuating and gradually worsen over years without significant diplopia (double vision is uncommon) [6,7]. Patients may have associated systemic features (“CPEO-plus”), such as cardiac conduction defects, retinopathy or endocrine abnormalities, but CPEO often presents without generalized symptoms initially [8].
Pathophysiology: CPEO is caused by defects in mtDNA or nuclear genes affecting oxidative phosphorylation, leading to reduced energy production and subsequent damage to muscle cells, particularly those in the extraocular muscles, which have very high mitochondrial content and low mutation thresholds [6]. Most cases result from large-scale mtDNA deletions or point mutations, whereas some are due to nuclear gene mutations. Common genetic mutations associated with CPEO include those in Polymerase Gamma, Catalytic Subunit (POLG), Polymerase Gamma 2, Accessory Subunit (POLG2), Thymidine Kinase 2, Mitochondrial (TK2), and optic Atrophy 1 (OPA1). Less commonly, the Mitochondrially Encoded tRNA Leucine 1 (MT-TL1) m.3243A>G mutation can also impair mitochondrial function, resulting in the accumulation of defective mitochondria and muscle degeneration [9].
Clinical Presentation: Patients typically have a slowly progressive, nonfluctuating eyelid ptosis and ophthalmoplegia that do not respond to edrophonium or immunotherapy [10]. On examination the levator function is reduced, and there is chronic external ophthalmoplegia without significant fatigability. In one series, CPEO patients first noticed ptosis (often asymmetric) with ophthalmoplegia developing later [10]. Diplopia can occur as ophthalmoplegia progresses but is initially uncommon. There may be associated dysphagia, neuropathy, or endocrinopathies if part of a “plus” syndrome. Blood lactate and pyruvate levels may be elevated after exercise. Genetic testing can confirm mtDNA deletions or mutations (or nuclear genes affecting mtDNA) underlying CPEO [8]. Importantly, nerve conduction and repetitive stimulation studies are normal, distinguishing CPEO from neuromuscular junction disorders [10,11]. EMG findings are nonspecific and typically show signs of myopathy, making EMG less useful for diagnosing CPEO. Muscle biopsy in CPEO classically shows ragged-red fibers (subsarcolemmal mitochondrial aggregates on Gomori trichrome stain) often exceeding the 2% threshold diagnostic for mitochondrial myopathy [12]. Mitochondrial genetic testing is considered the gold standard for diagnosing CPEO, with the most common mutation being a large-fragmentdeletion of mtDNA [7].
Management: There is no disease-modifying therapy for CPEO; treatment is supportive. Ptosis can be corrected surgically (levator advancement or frontalis sling) when eyelid droop significantly impairs vision [10]. Extraocular misalignment, if symptomatic, may be managed with prism glasses or strabismus surgery. Given cardiac conduction block is common in CPEO/Kearns-Sayre, patients require regular ECG monitoring and pacemaker implantation if high-grade atrioventricular block develops [10]. Endocrine complications (e.g. diabetes, hypothyroidism) should be managed per standard guidelines. Genetic counseling is appropriate once the diagnosis is confirmed [10,11].
Oculopharyngeal Muscular Dystrophy (OPMD)
OPMD is a late-onset autosomal dominant myopathy affecting primarily the eyelid and pharyngeal muscles [13]. It typically presents in mid-adulthood (mean age ~48 for ptosis and 50 for dysphagia). The classic triad is bilateral ptosis and progressive dysphagia; tongue weakness/atrophy is also common. Limb-girdle weakness may appear later in severe cases [13]. OPMD is a hereditary muscle disease caused by a polyalanine expansion in the PABPN1 gene [14,15]. Initial symptoms often include heavy eyelids and swallowing difficulty for solids, later involving liquids. Family history of similar symptoms is common due to autosomal dominant inheritance, although rare recessive forms exist [14,15]. Unlike myasthenia gravis, OPMD ptosis is insidious and nonfluctuating.
Pathophysiology: The pathogenic mechanism is a short GCN trinucleotide repeat expansion in Poly(A) Binding Protein Nuclear 1 (PABPN1), leading to an expanded polyalanine tract in the nuclear poly(A) binding protein [14]. Mutant PABPN1 protein forms intranuclear aggregates in muscle cells, disrupting RNA processing and leading to muscle fiber degeneration (especially in eyelid and pharyngeal muscles). Most patients carry 12-18 alanine repeats in one PABPN1allele. The aggregates are the pathologic hallmark and muscle biopsy shows dystrophic changes (fibers of varying size, rimmed vacuoles) [16]. Histology typically shows rimmed vacuoles and nuclear inclusions in atrophic fibers. Proximal limb muscles eventually become involved, but extraocular muscles are relatively spared, explaining the isolated ptosis and bulbar symptoms. The disease shows anticipation: larger expansions often cause earlier or more severe disease [17].
Clinical Presentation: Clinically, OPMD patients have a characteristic progression: slowly worsening bilateral ptosis (often symmetric) followed by bulbar weakness (dysphagia, nasal speech) and then proximal limb weakness [14,15]. Diplopia is uncommon. One report noted that dysphagia was the predominant complaint (years after ptosis) and exam showed unilateral eyelid ptosis and hoarseness [14]. A key clue is a family history of similar late-onset ptosis or dysphagia. Electrophysiology is normal for neuromuscular junction testing, shows a myopathic pattern, distinguishing it from the decremental response of MG. Creatine kinase is usually normal or mildly elevated. Muscle biopsy (if done) shows small, round fibers with rimmed vacuoles, but genetic testing for the PABPN1 expansion is diagnostic [18].
Management: There is no cure for OPMD. Treatment is supportive and symptomatic. Patients benefit from swallowing rehabilitation and dietary modifications to prevent aspiration. Cricopharyngeal myotomy or enteral feeding tubes may be considered for severe dysphagia [19]. Eyelid ptosis can be addressed with blepharoplasty or ptosis repair if it causes visual field obstruction. Exercise and physical therapy help maintain limb strength. Novel approaches (e.g. myostatin inhibition) are investigational. Close follow-up for weight loss and aspiration is important. Genetic counseling should be offered to the patient and family. As one review notes, “no definitive treatment” exists beyond these measures [19].
Myopathy associated with thyrotoxicosis
Thyrotoxicosis (usually Graves’ disease) can cause myopathy and characteristic orbital findings. Excess thyroid hormone accelerates metabolism and protein breakdown, leading to muscle fiber atrophy [20]. Patients with longstanding or severe hyperthyroidism may develop proximal muscle weakness and weight loss. In the orbits, Graves’ ophthalmopathy (thyroid eye disease) causes proptosis, eyelid retraction, conjunctival injection and restrictive ophthalmoplegia. Although classic Graves’ causes eyelid retraction, eyelid droop or diplopia can occur; their presence should prompt consideration of MG or concurrent myopathy. Importantly, ocular findings in Graves’ (exophthalmos, lid lag) are static, not fatigable [20,21], unlike MG. Systemic hyperthyroid features (tremor, heat intolerance, tachycardia) provide key clues.
Pathophysiology: Elevated thyroid hormones disturb muscle metabolism. Thyroxine impairs oxidative phosphorylation in muscle mitochondria and increases proteolysis via lysosomal enzymes. The net effect is accelerated protein catabolism and type 2 fiber atrophy [20]. Metabolic changes (increased cAMP signaling) create a hypermetabolic state that weakens muscle. These effects are reversible with normalization of thyroid hormone. Electrophysiologically, thyroid myopathy often shows a nonspecific myopathic pattern [20]. The precise mechanism is not fully understood, but reversible changes in Na+/K+ ATPase and altered membrane excitability have been proposed (explaining occasional MG-like decrement) [22]. Importantly, thyrotoxicosis can also unmask antibodies (e.g. to acetylcholine receptor) in susceptible individuals, further confusing the picture.
Clinical Presentation: Patients with thyrotoxic myopathy typically have signs of hyperthyroidism (weight loss, tachycardia, tremor, goiter) plus muscle weakness. The weakness is usually proximal (difficulty rising from chair, climbing stairs) [20]. Eyelid ptosis or ophthalmoparesis are rare but reported; when present, they may fluctuate, mimicking MG [22]. Serum CK is usually normal. Respiratory and bulbar muscles are rarely affected. Ocular exam classically shows upper lid retraction and lid lag; true ptosis is unusual. When ptosis or diplopia occur, thyroid ophthalmopathy is often evident (proptosis, extraocular muscle enlargement on imaging). Diagnosis is primarily clinical, supported by thyroid function tests (TSH, T3, T4) to assess thyroid status [21]. MRI or ultrasound of the orbits may reveal enlargement of extraocular muscles or orbital fat. EMG shows a myopathic pattern (short-duration, low-amplitude potentials) [20]. Importantly, Graves’ orbitopathy is static – ocular misalignment and lid position do not vary with fatigue [21].
Management: Treatment is aimed at the thyroid disease: antithyroid drugs (methimazole, PTU), beta-blockers for symptoms, and definitive therapy (radioiodine or thyroidectomy) as needed. Graves’ ophthalmopathy may require corticosteroids or orbital radiation/surgery for severe proptosis. Muscle weakness typically improves as thyroid levels normalize [20,21]. In summary, improvement of both ocular and muscular symptoms follows successful treatment of thyrotoxicosis [21]. No specific muscle-directed therapy is needed. Supportive care (physical therapy, eye patches for diplopia if present) can be used while thyroid treatment takes effect. It is also important to rule out true MG by antibody testing and decremental EMG; if those are negative, a thyroid cause should be sought [20,22].
Chronic ptosis: Chronic ptosis is a static drooping of the eyelid due to various causes. In adults, the most common is aponeurotic (involutional) ptosis from age-related levator aponeurosis dehiscence [23]. Other etiologies include myogenic (e.g. muscular dystrophy, oculopharyngeal dystrophy), neurogenic (oculomotor nerve palsy, Horner syndrome), mechanical (lid tumors or edema), congenital aponeurotic, and traumatic causes [23]. Unlike myasthenia, chronic ptosis is typically nonfluctuating and present for months to years.
Pathophysiology: Mechanisms vary by cause. In myogenic ptosis, the levator muscle itself is weak or dystrophic (e.g. muscular dystrophies or CPEO) Aponeurotic ptosis occurs when the levator aponeurosis attenuates or detaches. Neurogenic ptosis occurs when innervation is lost – e.g. a third nerve lesion causes ptosis plus pupil involvement and EOM deficits, while Horner syndrome (sympathetic denervation) causes mild ptosis with miosis and anhidrosis. Mechanical ptosis is due to an external load on the lid (e.g. a chalazion or dermatochalasis) or fibrosis. At the molecular level, neurogenic causes involve nerve injury, and myogenic causes involve intrinsic muscle pathology.
Clinical Presentation: Patients with chronic ptosis often describe a lifelong or progressive eyelid droop. Myogenic ptosis (aside from CPEO/OPMD) may be seen in childhood or early adulthood (e.g. myotonic dystrophy) and is usually symmetric. Neurogenic ptosis is usually unilateral and associated with other oculomotor findings: a CN III palsy has ptosis with “down and out” eye position and dilated pupil; Horner’s has ptosis with normal pupils on EOM testing and often a history of trauma or headache [24]. Mechanical ptosis is obvious if there is eyelid swelling or scarring, with preserved levator function. In all cases, levator excursion (measured with brow immobilized) helps distinguish aponeurotic ptosis (good levator function) from true muscle weakness (poor function) [23]. On exam, congenital ptosis is suggested by a poorly defined lid crease; paradoxical eyebrow elevation often occurs to compensate for ptosis. Fatigability tests (ice test, rest test) are negative in chronic ptosis of non-myasthenic cause.
Management: Treatment depends on cause. Aponeurotic (involutional) ptosis in an older patient is managed with surgical advancement of the levator aponeurosis (levator resection or tuck) if vision is impaired. Myogenic ptosis (e.g. dystrophy) may be approached similarly with blepharoplasty, though poor levator function may require frontalis slings. Neurogenic ptosis needs addressing the underlying lesion: for CN III palsy, this may mean aneurysm clipping or stroke rehab, while Horner’s syndrome may require imaging of the neck/chest to find a lesion. Mechanical ptosis is treated by removing the offending lesion (e.g. chalazion excision, ptosis repair after trauma). A general review notes that ptosis can have congenital or acquired causes and that management is tailored accordingly [23]. In all cases, ptosis surgery aims to improve the visual field and cosmesis; the risks and postoperative eyelid position must be discussed with the patient.
Functional (psychogenic) ptosis
Functional (psychogenic) ptosis is a conversion disorder in which eyelid droop occurs without structural pathology [25,26]. It is often precipitated by stress or psychological factors. True functional ptosis is rare but should be considered after excluding organic causes. Patients may present with unusual features, such as fixed ptosis that resolves with distraction or suggestion. Key clues are inconsistency (e.g. eyebrow (Müller’s muscle) may depress paradoxically during ptosis) and lack of objective abnormalities on testing.
Pathophysiology: The precise mechanisms are unclear. Psychologically, stressors or attention-seeking behavior are often identifiable, although the patient is not consciously faking the symptom [25]. Neurologically, functional disorders are thought to involve altered brain networks for movement and attention. In functional ptosis, voluntary pathways may override the levator palpebrae superioris muscle in response to psychologic cues, although the precise mechanism is not fully understood. Unlike organic ptosis, there is no structural damage to muscle or nerve.
Clinical Presentation: Functional ptosis often appears abruptly or with atypical features. In case reports, patients have isolated, unilateral or bilateral ptosis that fluctuates or resolves with certain maneuvers [26,27]. For example, one teenager with conversion ptosis had normal eyelid strength on detailed exam and eyebrow depression on the affected side [26]. The ice test and edrophonium test are typically negative. Electrophysiological studies (single-fiber EMG, repetitive stimulation) are normal [27]. Psychosocial history often reveals stressors (e.g. illness of a family member) [27]. A useful hallmark is that symptoms improve under suggestive conditions: one case resolved with placebo injection, confirming the functional nature [27].
Management: Functional ptosis is managed by first confirming no organic disease. Emphasis is on patient education and reassurance, avoiding unnecessary medications or surgeries. Once organic causes are excluded, a gentle explanation of the diagnosis is provided. Early involvement of a psychiatrist or psychologist is important. Cognitive-behavioral therapy and physical therapy (with focus on “retraining” normal movement) can be effective. In reported cases, symptoms often remit with psychological intervention [25,27]. With prompt recognition and support, recovery is common in children and adolescents. Indeed, studies show full recovery in >80% of pediatric conversion disorder cases with timely intervention [26]. No immunotherapy or surgery is indicated. Supportive measures include eye lubrication and eyelid taping if needed for function until recovery. It is critical to follow-up on any underlying stressors or psychiatric diagnoses (e.g. anxiety, depression) as part of holistic management.
Conclusion
OMG can be easily confused with a variety of other conditions that cause ptosis and ophthalmoplegia. Clues such as symmetry vs. asymmetry, presence of diplopia or systemic signs, electrophysiologic patterns, and specific laboratory findings help distinguish these mimics. For example, a static course and genetic/biopsy findings point to CPEO, while dysphagia and PABPN1 gene testing indicate OPMD. Conversely, fluctuating eyelid weakness and positive MG serology remain the sine qua non of OMG. Early consideration of these alternatives – especially in seronegative or treatment-refractory cases – is essential to avoid years of misdiagnosis. Recent advances in genetic testing and neurophysiology improve diagnostic accuracy, ensuring patients receive appropriate therapy rather than unnecessary immunotherapy.
References
- Evoli A, Iorio R. Controversies in Ocular Myasthenia Gravis. Front Neurol. 2020;11:605902.
- Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16:459-467.
- Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet. 2001;357:2122-2128.
- Harrison P, Barton J, Winkel A. Chronic mimics of myasthenia gravis: a retrospective case series. Neuromuscul Disord. 2023;33:250-256.
- Manini A, Caporali L, Meneri M, et al. Case Report: Rare Homozygous RNASEH1 Mutations Associated With Adult-Onset Mitochondrial Encephalomyopathy and Multiple Mitochondrial DNA Deletions. Front Genet. 2022;13:906667.
- Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain Sci. 2024;14:135.
- Feng Z, Lai R, Wei J, et al. Have one’s view of the important overshadowed by the trivial: chronic progressive external ophthalmoplegia combined with unilateral facial nerve injury: a case report and literature review. Front Neurol. 2024;14:1268053.
- Goldstein A, Falk MJ. Single Large-Scale Mitochondrial DNA Deletion Syndromes. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2025.
- Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2025.
- Wabbels B, Ali N, Kunz WS, et al. Chronisch-progressive externe Ophthalmoplegie und Kearns-Sayre-Syndrom: Interdisziplinäre Diagnostik und Therapie. Ophthalmologe. 2008;105:550-556.
- Seemongal-Dass RV, Gracen DJ, Seemongal-Dass RR, et al. Chronic Progressive External Ophthalmoplegia: A Case Report. Cureus. 2025;17:e77149.
- Lv H, Qu Q, Liu H, et al. Clinical, neuroelectrophysiological and muscular pathological analysis of chronic progressive external ophthalmoplegia. Exp Ther Med. 2020;20:1770-1774.
- Brais B. Oculopharyngeal muscular dystrophy. Handb Clin Neurol. 2011;101:181-192.
- Werling S, Schrank B, Eckardt AJ, et al. Oculopharyngeal muscular dystrophy as a rare cause of dysphagia. Ann Gastroenterol. 2015;28:291-293.
- Goyal NA, Mozaffar T, Chui LA. Oculopharyngeal Muscular Dystrophy, an Often Misdiagnosed Neuromuscular Disorder: A Southern California Experience. J Clin Neuromuscul Dis. 2019;21:61-68.
- Aryani O, Akbari M, Aghsaei-Fard M, et al. Oculopharyngeal muscular dystrophy misdiagnosed as myasthenia gravis: Case report and review of literature. Iran J Neurol. 2017;16:98-99.
- Harper PS, Harley HG, Reardon W, et al. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet. 1992;51:10-16.
- Trollet C, Boulinguiez A, Roth F, et al. Oculopharyngeal Muscular Dystrophy. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2025.
- Chen AW, Wu SL, Cheng WL, et al. Dysphagia with fatal choking in oculopharyngeal muscular dystrophy: Case report. Medicine (Baltimore). 2018;97:e12935.
- Kim TJ, Lee HS, Shin JY, et al. A case of thyrotoxic myopathy with extreme type 2 fiber predominance. Exp Neurobiol. 2013;22:232-234.
- Smółka K, Perenc L, Pelc J, et al. Thyrotoxic Myopathy with Nonspecific Ophthalmopathy in a Two-Year-Old Child: Case Report and Literature Review. J Clin Med. 2024;13:6180.
- Tsuda E, Imai T, Matsumura A, et al. Thyrotoxic myopathy mimicking myasthenic syndrome associated with thymic hyperplasia. Intern Med. 2008;47:445-447.
- Finsterer J. Ptosis: causes, presentation, and management. Aesthetic Plast Surg. 2003;27:193-204.
- A review of ocular myasthenia gravis and its differential diagnoses. J Med Optom [Internet]. Available from: https://journalofmedicaloptometry.com/volume-3-issue-1/a-review-of-ocular-myasthenia-gravis-and-its-differential-diagnoses/
- Bagheri A, Abbasnia E, Pakravan M, et al. Psychogenic unilateral pseudoptosis. Ophthalmic Plast Reconstr Surg. 2015;31:e55-e57.
- Akçay A, Yılmaz S, Serdaroğlu G, et al. A rare presentation of conversion disorder: palpebral ptosis. J Pediatr Res. 2014;1:33-35.
- Peer Mohamed BA, Patil SG. Psychogenic unilateral pseudoptosis. Pediatr Neurol. 2009;41:364-366.
