
Open Access, Volume 11
Hypoplastic/aplastic gallbladder vs chronic contracted gallbladders: A diagnostic challenge
Akhavan V Ryan, BS1*; Saed Khaled, MD2; Erdman Dan, BS1; Young Jason, BS1; O’Neal Andrew, MD, FACS3
1St. George’s University, USA.
2Larkin Community Hospital, USA.
3Delray Medical Center, USA.
Ryan Akhavan
St. George’s University School of Medicine, St. George’s, Grenada, USA.
Email: rakhavan@sgu.edu
Received : June 11, 2025,
Accepted : July 17, 2025
Published : July 31, 2025,
Archived : www.jclinmedcasereports.com
Abstract
Congenital gallbladder aplasia and maldevelopment is a rare biliary tree anomaly, accounting for somewhere between 0.01% to 0.065% of the population. Some patients may develop symptoms of biliary tree diseases, including right-upper quadrant abdominal pain, nausea, vomiting, jaundice and fever, which can cause diagnostic challenges. We followed a 79-year-old female that initially presented with urosepsis followed by abnormal liver enzymes. Imaging studies, including CT scan, ERCP, and HIDA scan, demonstrated extrahepatic biliary dilation as well as conflicting findings, ranging from a contracted gallbladder to a non-definitely visualized gallbladder. Our patient was scheduled for a cholecystectomy. Intraoperatively, direct diagnosis of gallbladder aplasia was confirmed. The preoperative diagnosis of a contracted acalculous chronic cholecystitis associated with inconclusive imaging results of the gallbladder anatomy may indicate congenital gallbladder aplasia. Given the difficulty of preoperative diagnosis, having a congenital abnormality as a differential diagnosis will also help in the possible intraoperative discovery of this rare pathology and avoidance of unnecessary surgical morbidity and mortality. This case report will focus on delineating between the various modalities of diagnosing and identifying congenital gallbladder aplasia, varying from going in laparoscopically or solely by imaging studies, including abdominal ultrasound and CT scans.
Keywords: Congenital aplasia; Biliary atresia; Cholecystitis; Congenital abnormality; Case report.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Ryan AV (2025)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Ryan AV, Khaled S, Dan E, Jason Y, Andrew ON. Hypoplastic/aplastic gallbladder vs chronic contracted gallbladders: A diagnostic challenge. Open J Clin Med Case Rep. 2025; 2370.
Background
The biliary system, known as the biliary tract, is a system of organs and ducts that include the liver, gallbladder and bile ducts [1]. The gallbladder and associated organs are derived from the primitive endoderm during the 4th week of development. The structure known as the hepatic diverticulum gives rise to the bile ducts, which then connects with the midgut. The gallbladder and the cystic duct then form from a ventral outgrowth of the bile duct [2].
There are several congenital gallbladder anomalies, including gallbladder aplasia, deformation, atresia, and duplication, amongst others. Congenital aplasia of the gallbladder is a rare condition that occurs in 0.01% to 0.065% of the population [3]. Females are more commonly affected than males (3:1 ratio), which typically present in the second or third decade of life [4]. Many cases of familial aplasia have been reported, suggesting a hereditary link [5].
The majority of patients with agenesis of the gallbladder do not suffer clinically significant complications from cystic aplasia. However, it is estimated that 25% of patients with Gallbladder agenesis will present with biliary tree pathologies, necessitating that clinicians are able to quickly recognize and adjust for these conditions. Gallbladder agenesis can be associated with other anomalies, such as Klippel Feil syndrome, malrotation of the gut, and horseshoe kidney [6].
In the patient population with Gallbladder agenesis, approximately 40 to 65% of patients display the typical symptoms of cholelithiasis, including right-upper quadrant pain, fever, nausea, vomiting and jaundice [7]. Biliary aplasia can pose difficulty in both the diagnosis and surgical management, especially during laparoscopic cholecystectomy or MRCP, where the missing gallbladder can be mistaken for an outpouching or a cyst. Gallbladder aplasia is a rare occurrence and not widely reported in the literature. Gallbladder aplasia may be misdiagnosed with other conditions.
A chronic contracted gallbladder refers to a permanently atrophied gallbladder as a result of repeated episodes of inflammation. The incidence is greater in older women as estrogen increases biliary cholesterol secretion causing cholesterol supersaturation of bile [8]. Inflammation most commonly comes in the form of chronic cholecystitis, often with the addition of gallstones [9]. As a result of repeated inflammation, the gallbladder becomes inelastic, with scarred thickened walls. This chronic cellular change is postulated to increase the risk of malignancy due to continued inflammation leading to histological changes including, metaplasia, dysplasia and hyperplasia [10,11]. Patients may be asymptomatic but often experience symptoms of right upper quadrant abdominal pain, nausea, vomiting or fatty food intolerance. Gold standard diagnostic modality includes Ultrasound which can demonstrate full or partial wall thickening as well as atrophy [12]. Patients who are symptomatic are good surgical candidates for laparoscopic cholecystectomy. The diagnoses of chronic contracted gallbladder may mimic congenital anomalies of the gallbladder which may pose difficulty and require intraoperative diagnosis. Special attention must be made to differentiate these two pathologies intraoperatively in order to mitigate unnecessary surgical predicaments.
The preoperative diagnosis of Gallbladder agenesis has shown to be extremely difficult, which has led to most definitive diagnoses to be made during intraoperative investigation [13]. It is possible that clinicians or radiologists that are not familiar with identifying these abnormalities refrain from documenting what they observe [13]. Studies have shown that Magnetic Resonance Cholangiopancreatography (MRCP) is the gold standard tool to establish Gallbladder agenesis, as it allows a biliary cartography and should be used preoperatively to complement inconclusive ultrasounds [13]. When preoperative diagnosis is not achieved, undergoing laparoscopic cholecystectomy for congenital gallbladder aplasia can give rise to complications. In cases of unclear intraoperative anatomy, it is recommended to abort the procedure rather than to find the gallbladder due to increased risk of iatrogenic injury [14].
We hope our case report provides information and guidance on how to diagnose and manage persons with congenital Gallbladder anomalies. By reporting our patient’s history, clinical course and outcomes, we hope to enhance the understanding and further guidelines on how to manage these patients. In our case report, we followed a 79-year-old female patient who was found to have Gallbladder aplasia on imaging, which was confirmed intraoperatively and later by pathologic examination.
Case Presentation
The patient, a 79-year-old female, was initially admitted to the hospital on June 24th with complaints of worsening right knee pain and a confirmed Urinary Tract Infection (UTI). During her stay, on June 26th, she developed a fever of 39.6°C (103.3°F), and her White Blood Cell (WBC) count rose to 20,000 per microliter of blood, indicating a possible infection or sepsis. Her Liver Function Tests (LFTs) were also abnormal, with an Alkaline Phosphatase (ALP) level of 205 IU/L, Alanine Transaminase (ALT) of 67 IU/L, and Aspartate Transaminase (AST) of 84 IU/L.0
These abnormal results, in the background of sepsis, prompted further investigation for a biliary etiology as a source of sepsis.
On June 27, 2024, a CT scan of the abdomen was performed, revealing significant findings. The report described ‘‘biliary ductal obstruction at the ampulla with severe biliary ductal dilatation.’’ Specifically, the common bile duct was measured at 1.6 cm in diameter, and the common hepatic duct was 2.3 cm, indicating a notable obstruction. Additionally, the scan noted ‘‘fluid and adjacent duodenal sweep but no soft tissue mass.’’
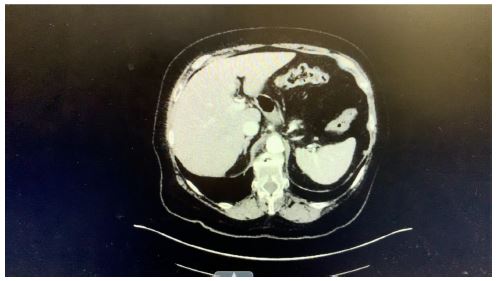
Figure 1: CT scan of the abdomen.
To further investigate the biliary system, a Hepatobiliary Iminodiacetic Acid (HIDA) scan was conducted on July 9, 2024. The HIDA scan showed «prompt uptake and excretion in the liver,» with visualization of the bile ducts within 5-10 minutes. The scan revealed «nonvisualization of the gallbladder,» that is considered a positive finding for acute cholecystitis which also is a finding for a patient with congenital aplasia and will be a reasonable finding for a post-cholecystectomy patient.
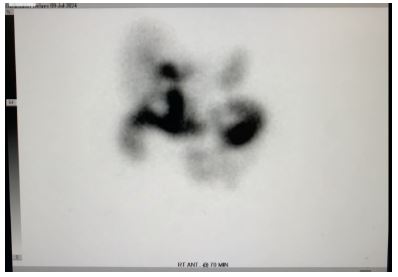
Figure 2: HIDA scan.
An Endoscopic Retrograde Cholangiopancreatography (ERCP) performed on July 10, 2024, as a possible diagnostic and therapeutic management for obstructive jaundice. The ERCP findings indicated a large unclear tubular structure where the cystic duct should be, with no identifiable gallbladder. The contrast study confirmed the possibility of an obstructed gallbladder versus gallbladder aplasia. The ERCP highlighted significant dilation of the bile ducts, consistent with the earlier CT findings.
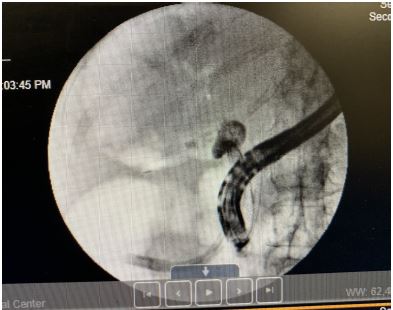
Figure 3: Endoscopic retrograde cholangiopancreatography (ERCP).
Given our patient’s presentation, as well as imaging findings, our preliminary diagnosis of acute on chronic cholecystitis warranted laparoscopic cholecystectomy, done on July 3rd, to definitively manage the disease and prevent future biliary tree obstruction. Intraoperatively, direct evidence of gallbladder aplasia was confirmed. The initial assumption of the surgical team was an «extremely contracted gallbladder and fused with the common bile duct,» making separation impossible. On further exploration, a “tubular structure» where the cystic duct should have been, was identified with a complete absence of a gallbladder. This absence was consistent with the imaging results and the ERCP findings.
The patient’s initial presentation, including symptoms of sepsis and elevated inflammatory markers, was complicated by the rare finding of congenital gallbladder agenesis. The imaging findings from the CT scan and HIDA scan, along with the ERCP results and surgical exploration, collectively confirmed the diagnosis. The CT scan showed biliary ductal obstruction and dilation, while the HIDA scan’s nonvisualization of the gallbladder and the ERCP’s findings of a tubular structure without a gallbladder were consistent with the congenital aplasia. Given the patient has a clinical and paraclinical picture of obstructive jaundice with acute cholangitis, the team decided to conduct an ERCP and circumvent the use of an MRCP to further aid in diagnosis and therapy.
Intraoperative findings corroborated the imaging results, revealing a fused and contracted gallbladder and an aberrant anatomical structure where the cystic duct should have been. This comprehensive diagnostic approach highlighted the rarity of the patient’s condition and the importance of accurate imaging and surgical evaluation in confirming congenital anomalies.
Overall, the case underscores the significance of integrating multiple diagnostic modalities—CT scan, HIDA scan, ERCP, and surgical findings—to accurately diagnose and manage complex biliary conditions such as congenital gallbladder agenesis. The detailed imaging and surgical evidence provided a clear and consistent diagnosis, elucidating the patient’s rare and challenging condition.
Discussion
There are a myriad of inflammatory pathologies of the biliary tree, yet all these conditions share many clinical features. Many cases may present with right-upper quadrant abdominal pain, nausea, and intolerance to fatty foods, however this may mimic other aberrations of the regional parenchyma or peritoneum, including cholelithiasis, cholecystitis, or choledocholelithiasis [13]. However, clinical presentation, diagnostic criteria, and treatment methods tend to become more complicated when encountering similar pathologies with embryological origins, as was the case with our patient with evidence of gallbladder aplasia.
Other case reports delineating cases of gallbladder aplasia have been conducted, showing its relation to similar gallbladder pathologies. Thant et. al showed that diagnosis occurred during a laparoscopy for a presumed diagnosis for cholecystitis. Similar to our case study, the patient presented with right-upper quadrant pain with a right-upper quadrant abdominal ultrasound displaying difficult visualization of the gallbladder and calcific stones. The patient was then given antibiotics with eventual cholecystectomy 2 weeks later. Intraoperatively, no gallbladder was seen, and the patient conducted an outpatient MRCP, which showed no gallbladder or cystic duct, confirming the diagnosis of congenital gallbladder aplasia [14]. MRCP is usually the preferred method for confirmation of gallbladder aplasia, as it is non-invasive and allows for direct examination of the biliary tract.
Another study by Takano et. al showed a pediatric patient presenting with fever, fatigue, and elevated LFTs (AST of 406 IU/L, ALT of 227 IU/L) and elevated alkaline phosphatase (798 IU/L). What was particularly unique about this case study was that the patient was diagnosed using only imaging modalities, including an abdominal ultrasound and MRCP, which revealed an absent gallbladder, and Drip Infusion Cholecystocholangiography with Computed Tomography (DIC-CT) [15]. DIC-CT is particularly efficient, as it uses a radiopaque dye that accumulates in the gallbladder and bile ducts and can confirm the absence of the gallbladder [15]. Laparoscopy and laparotomy were not completed as in the previous case described, and acute cholecystitis was ruled out given the lack of recurrent right-upper quadrant abdominal pain.
In this case, anomalous formation of this patient’s hepatobiliary system presented under the guise of gallbladder inflammation. Symptomatology is an unequivocal indicator as to the proper methodology of treatment, and can guide the direction of care in complex cases. As our patient experienced abnormal liver function tests in addition to evidence of biliary tract anomalies, surgical intervention may have indeed been warranted. The literature states that certain biliary pathologies indicating biliary duct dilation, including choledocholelithiasis, may not be as easily detected solely with imaging modalities such as MRCP and DIC-CT [15]. This indicates the need for ERCP or potential surgical evaluation, such as laparoscopy, to further confirm the diagnosis and delineates the necessity for complete evaluation of the patient’s concerns and symptoms.
Patients presenting with symptomatic biliary tree inflammation with concurrent anomalous gallbladder anatomy often have similar presentation, regardless of whether or not the anomaly is acquired, as in the case of contracted gallbladder or congenital gallbladder aplasia [16]. However, the conditions often differ in terms of surgical intervention and long-term management [4]. Distinguishing biliary tree inflammation in the context of contracted gallbladder from agenesis is crucial. They often present similarly on imaging and even present with similar History of Presenting Illnesses (HPI), with existing literature on cases where a contracted gallbladder on imaging was found to be agenesis upon visualization laparoscopically [16]. Much of the diagnostic ambiguity arises from the presence of retrohepatic and ectopic gallbladder as a source of inconclusive imaging. The extent of clinical overlap necessitates a greater awareness for gallbladder agenesis to prevent unnecessary surgical intervention, and by extension, possible iatrogenic injury and a protracted recovery.
This is a case that clearly illustrates the utility for distinguishing agenesis from contracted gallbladder preoperatively. Here, contracted gallbladder was erroneously identified on imaging preoperatively, resulting in the abortion of surgical intervention upon visualization of an aplastic gallbladder. In this case, a comprehensive work-up was done, with many indications of a possible contracted gallbladder. In this particular case, a 63-year-old female presented with RUQ pain radiating to the back and a remote history of parathyroidectomy [16].
Most cases of gallbladder agenesis appear as a contracted or shrunken gallbladder, which further necessitates the need to go in laparoscopically and confirm the diagnosis [3].
Christophoros et. al showed a case report of a 62-year-old male who presented to the clinic with the typical signs of right upper-quadrant abdominal pain, nausea, and vomiting. When conducting a laparoscopy, the gallbladder could not be properly identified [3]. While imaging such as ultrasound or CT can be conducted to further identify the pathophysiology of similar conditions relating to the gallbladder, preoperative diagnosis may not be sufficient with such modalities, and further investigation may be required. This is similar to our patient whose prediagnosis was also contracted gallbladder, whereby a congenital anomaly was later identified through laparoscopy. The current research lacks proper understanding of delineating between contracted gallbladder and congenital gallbladder aplasia, and further highlights the importance of proper management to properly attach the correct diagnosis. This case further emphasizes the need to refine diagnostic algorithms, as this patient had a long history of right upper-quadrant pain, making it easy to reasonably assume the patient had fibrotic changes to the gallbladder resulting from inflammation. Additionally, the comparative greater prevalence of contracted gallbladder often leaves indications of gallbladder aplasia overlooked.
Conclusion
Delineating between congenital gallbladder aplasia and a chronic contracted gallbladder can present a diagnostic challenge, as they both present similarly and have overlapping clinical features and imaging findings. Our case illustrates the importance of a thorough and integrated diagnostic approach, utilizing advanced imaging techniques such as CT scans, HIDA scans, and ERCP, alongside surgical evaluation, to confirm the diagnosis of gallbladder aplasia. While the literature generally states to complete an MRCP for cases of contracted gallbladder or congenital gallbladder anomalies, we elected to bypass the MRCP and conduct an ERCP given the patient’s obstructive jaundice and risk factors for cholangitis. This case underscores the need for enhanced awareness of having congenital gallbladder etiologies as a differential diagnosis, as misdiagnosis can lead to inappropriate management and increased morbidity. Further research and documentation of such cases will be crucial in refining our approaches and ensuring that clinicians are equipped to recognize and manage these complex presentations effectively.
References
- Hopkins Medicine. Biliary System Anatomy and Functions. Johns Hopkins Medicine. 2024.
- Standring S. Gray’s Anatomy: The Anatomical Basis of Clinical Practice. Elsevier Health Sciences. 2016.
- Kosmidis CS, Koimtzis GD, Kosmidou MS, Ieridou F, Koletsa T, Zarampouka KT, Georgakoudi E, Kesisoglou I. Gallbladder hypoplasia, a congenital abnormality of the gallbladder: a case report. Am J Case Rep. 2017; 18: 1320–1324.
- Kasi PM, Ramirez R, Rogal SS, Littleton K, Fasanella KE. Gallbladder agenesis. Case Rep Gastroenterol. 2011; 5: 654–662.
- Gallbladder agenesis. ScienceDirect. 2024.
- Gallbladder agenesis. Radiopaedia. 2024.
- Peloponissios N, Gillet M, Cavin R, Halkic N. Agenesis of the gallbladder: a dangerously misdiagnosed malformation. World J Gastroenterol. 2005; 11: 6228–6231.
- Sun H, Tang H, Jiang S, Zeng L, Chen EQ, Zhou TY, Wang YJ. Gender and metabolic differences of gallstone diseases. World J Gastroenterol. 2009; 15: 1886–1891.
- Park YH. Contracted chronic cholecystitis with gallstone: a case report. Clin Ultrasound. 2018; 3: 24–27.
- Arora VK, Kumar S, Singh N, Bhatia A. Intraoperative bile cytology of the dysplasia-carcinoma in situ sequence of gallbladder carcinoma. Cancer. 2005; 105: 277–281.
- Duarte I, Llanos O, Domke H, Harz C, Valdivieso V. Metaplasia and precursor lesions of gallbladder carcinoma: frequency, distribution, and probability of detection in routine histologic samples. Cancer. 1993; 72: 1878–1884.
- Bani-Hani KE. Agenesis of the gallbladder: difficulties in management. J Gastroenterol Hepatol. 2005; 20: 671–675.
- Peloponissios N, Gillet M, Cavin R, Halkic N. Agenesis of the gallbladder: a dangerously misdiagnosed malformation. World J Gastroenterol. 2005; 11: 6228–6231.
- Phyu CT, Gupta A. Agenesis of gallbladder—an unexpected absence in laparoscopy. J Surg Case Rep. 2023; 2023: rjad214.
- Takano Y, Hoshino M, Iriyama S, et al. Gallbladder agenesis with hepatic impairment: a case report. BMC Pediatr. 2018; 18: 360.
- Anderson K, Roland AL, Miller MP, Foretia DA. Beware of the shrunken gallbladder: case report of intraoperatively diagnosed gallbladder agenesis. Int J Surg Case Rep. 2022; 98: 107588.
