
Open Access, Volume 11
The role of increased plasma K+ level in the accelerated osteoporosis induced by synthetic glucocorticoids owning decreased mineralocorticoid effects
Sándor Sipka1*; Andrea Nagy1; József Szentmiklósi2; Sándor Sipka Jr3; Zsolt Bodnár4; Tünde Tarr1
1Division of Clinical Immunology, Faculty of Medicine, University of Debrecen, Hungary.
2Department of Pharmacology, Faculty of Medicine, University of Debrecen, Hungary.
3Division of Cardiology, Department of Cardiology, Faculty of Medicine, University of Debrecen, Hungary.
4Department of Surgery, Letterkenny, Ireland.
Sándor Sipka
Division of Clinical Immunology, Faculty of Medicine, University of Debrecen, Móricz Zsigmond str. 22. 4032 Debrecen, Hungary.
Tel: +36 (52) 255-218, Fax: +36 (52) 255-218;
Email: sipka.sandor45@gmail.com
Received : December 11, 2024,
Accepted : January 09, 2025
Published : January 15, 2025,
Archived : www.jclinmedcasereports.com
Abstract
Background: Glucocorticoid Drugs (GS) (synthetic glucocorticoids) are highly selective glucocorticoid receptor agonists but have minimal mineralocorticoid activity. They are mainly used for the therapy of allergic and autoimmune diseases as anti-inflammatory and immunosuppressant agents. However, they often cause negative side effects, such as osteoporosis leading to bone fractures.
Objective: This study aims to demonstrate those main processes that can induce imbalances in mineral homeostasis and cause osteoporosis during GS therapy from the aspect of elevated plasma level of K+. Dexamethasone (DXM), as a strong representative of GS was chosen as the model molecule of effects used in severe diseases generating strong danger signals for the organism.
Results: In Model 1, the biochemical processes of classical stress reactions are presented in a synthesized way. Model 2 shows the disturbances induced by DXM leading to high plasma level of K+. In Model 3, those main extracellular processes are synthesized which are involved in the development of serious osteoporosis induced by CS therapy.
Conclusion: This is the first work emphasizing the crucial role of elevated plasma K+ level in the development of severe osteoporosis induced by synthetic glucocorticoids.
Keywords: Adenosine triphosphate; Adenosine; Stress hormones; Synthetic glucocorticoids; Osteoporosis.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Sipka S (2025)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Sipka S, Nagy A, Szentmiklosi J, Sipka Jr S, Bodnar Z, Tarr T. The role of increased plasma K+ level in the accelerated osteoporosis induced by synthetic glucocorticoids owning decreased mineralocorticoid effects. Open J Clin Med Case Rep. 2025; 2317.
Introduction
Osteoporosis, the most frequent form of the metabolic bone-characterized disease, is a skeletal disorder characterized by compromised bone strength predisposing a person to an increased risk of fracture [1]. Although osteoporosis can occur at any age and in both genders, it typically is an age-related disease that more frequently affects women than men [2]. However, Glucocorticoid-Induced Osteoporosis (GIOP) is the most common form of secondary osteoporosis [3]. Synthetic Glucocorticoids (GS) are used in a wide variety of disorders, including autoimmune, pulmonary and gastrointestinal diseases, as well as in patients following organ transplantation and with malignancies. Although the indications for glucocorticoids in these various conditions are clear, their use is fraught with a host of potential side effects. One organ system that has the potential to be profoundly affected by glucocorticoids is the skeleton [3]. The natural glucocorticoid molecules are steroid hormones derived from cholesterol and are secreted by the adrenal cortex. Their role is essential for homeostasis and many vital functions, such as growth, reproduction, immune and stress responses, behavior, and cell proliferation. Their cellular actions are mediated via the intracellular Glucocorticoid (GR) and Mineralocorticoid (MR) receptors. Their productions are under the feedback control of Corticotrophin-Releasing Hormone (CHR) from brain nuclei and Adrenocortical Hormone (ACTH) from the anterior pituitary gland, stimulating the production of cortisol and aldosterone from the cortex of the adrenal gland, creating the Hypothalamus-Pituitary-Adrenal gland (HPA) axis. When danger signals impose stressful stimuli, the stress response starts rapidly, within seconds. It is characterized by augmented secretion of norepinephrine (noradrenaline) and epinephrine (adrenaline) from the elements of the peripheral sympathetic nervous system followed by activation of the HPA axis, which induces the increased release of ACTH involving the secretion of glucocorticoids (cortisol) and mineralocorticoids (aldosterone) from the cortex of adrenal glands [4].
Intracellularly, the mitochondria are the key components of the stress response, regulated by Ca2+, ATP, Reactive Oxygen Species (ROS) metabolism, and the intensity of apoptosis [5]. We recently created a new synthesized model named “extracellular ATP-adenosine danger cycle” [6]. Depending on the strength of various danger/stress signals, quantitative and qualitative changes are taking place in the cells: a) transient and repairable loss of ATP; b) “apoptosis” with irreversible loss of ATP; c) “necrosis”, with total loss of ATP and viability, but induction of secondary inflammation by the necrotic cells. Both ATP and adenosine have intracellular “metabolic” and extracellular “signal” functions. Extracellular ATP molecules mediate the signals of defense for the cells by emitting reactive messenger molecules (cytokines, ROS, H2O2). In contrast, extracellular adenosine produced from ATP serves the survival of cells and the regeneration of ATP pools [6].
Chemical modifications increased the anti-inflammatory and immunosuppressant effects of cortisol, resulting in the development of the family of synthetic Glucocorticoids (GS). The history of corticosteroid therapy started with Cortisone in 1948, followed by Hydrocortisone (1950), Prednisone (1954), Prednisolone (1954), Fluorocortisone (1954), Triamcinolone (1956), Methyl-prednisolone (1957), Dexamethasone (1958) [7]. However, the augmentation of glucocorticoid activity in these molecules led to the loss of their mineralocorticoid activity. Table 1 shows the various synthetic GS drugs with their glucose and mineralocorticoid potencies. Dexamethasone (DXM) is the strongest representative of these molecules, so we used it as the “model molecule” in the Figures.
Table 1: Relative potency and mineralocorticoid activity of commonly used glucocorticoids.
| Relative potency | Daily dose (mg) | |||
|---|---|---|---|---|
| Steroids | Glucocorticoids | Mineralocorticoids | ||
| Short-Acting | Cortisol | 1.0 | 1.0 | 30.0 |
| Cortisone | 0.8 | 0.8 | 37.5 | |
| Prednisone | 4.0 | 0.0 | 7.5 | |
| Prednisolone | 4.0 | 0.8 | 7.5 | |
| Intermediate-Acting | Triamcinolone | 5.0 | 0.0 | 6.0 |
| Long-Acting | Bethamethasone | 25.0 | 0.0 | 1.2 |
| Dexamethasone | 30.0 | 0.0 | 1.2 | |
Textbook of Internal Medicine, ed. Kelly WN. p: 2192,1989
The mechanisms of GIOP are summarized as follows: rapid decrease in bone formation, increase in bone resorption, decreased gut absorption of calcium, increased urinary excretion of calcium, secondary hyperparathyroidism, decreased muscle mass, decreased bone matrix, suppression of gonadal function and suppression of adrenal androgen secretion [8]. In the acceleration of osteoporosis and bone fractures, glucocorticoids have strong direct effects on the osteocytes (decreasing function), osteoblasts (decreasing of differentiation), and osteoclasts (stimulation of their genesis and function) in the bones. In addition, they increase the renal excretion of Ca2+ and block its intestinal reabsorption by vitamin D. The inhibitions of the production of insulin-like growth factor (stimulating bone formation) and the synthesis of sex hormones (inhibiting bone resorption) and the appearance of myopathy all contribute to the increased risk of fractions [3]. However, we suppose that in the processes leading to secondary hyperparathyroidism, the decreased mineralocorticoid activity of synthetic glucocorticoids plays a central role. One aim of this work is to elucidate this idea in detail. Furthermore, we aimed to study GIOP from the aspect of “natural” stress response when the increased production of cortisol and aldosterone strongly coordinates the metabolic answers of defense against the danger signals but cooperates with Parathyroid Hormone (PTH), too.
The current review presents two types of new models: a) The interactions between the ATP-adenosine-related intra- and extracellular molecules and the basic stress hormones during „natural” stress situations (Model 1); b) The crucial role of deficient mineralocorticoid activity of GS drugs in the acceleration of osteoporosis coupled to stress responses (Model 2 & Model 3).
Model 1: “Extracellular ATP-adenosine danger cycle” and the peripheral interactions of main stress hormones
This composition is based on our earlier work describing the elements of the “Extracellular ATP-adenosine danger cycle” [6] during “natural” stress situations completed with the consideration of Parathyroid Hormone (PTH), too. The various danger signals induce “stress responses” by activating specific elements of central and peripheral nervous systems, activating the HPA axis, resulting in ACTH, cortisol and aldosterone productions. In addition, great amounts of adrenaline are produced by the medulla of the adrenal gland, inducing strong release of ATP and other messenger molecules from the cells acting via P2R purinergic receptors and serving the defense of organisms by “short term”, functionally “whip” effects. In parallel, great amounts of Adenosine (Ado) are produced from ATP by the endonucleotidases acting via P1R purinergic receptors, preventing the overstimulation of cells and supporting their survival. In a later phase, both intracellular ATP and Ado take part in regenerating energy pools of ATP. Extracellular Ado also has anti-inflammatory and immunosuppressive effects. Our suggestion was to build it in Model 1 (Figure 1), namely, the chance that very high doses of Ado can still increase the stress response by induction of ACTH from the anterior pituitary gland [9]. Adrenaline is metabolized (deaminated) by Monoamine Oxidase (MAO) [10], and Adenosine Deaminase (ADA) transforms inosine from Ado [11]. Cortisol has a central role in the processes by stimulation of adrenaline production and controlling the production of “ACTH” via a negative “feedback regulation” [4]. Cortisol has anti-inflammatory and immunosuppressive capabilities. Besides, cortisol can support aldosterone in the reabsorption of Na+ by the kidney, especially during stress response, as it has a slight mineralocorticoid potency, too [4]. However, in stressful situations, these processes can still be multiplied and strengthened, involving, among others, the increased production of Parathyroid Hormone (PTH), which is initiated by the elevated aldosterone level [12]. These extra and intracellular situations show that already weak forms of stress responses can accelerate bone resorption Figure 1 Other forms of endocrine disturbances can also occur in stress, e.g., insulin resistance [13].
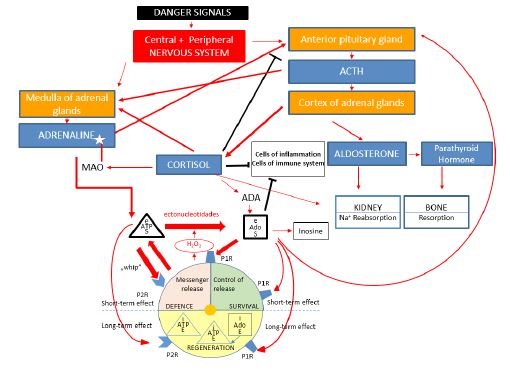
Figure 1: Extracellular ATP-adenosine danger cycle” and the peripheral interactions of main stress hormones.
ACTH: Adrenocorticoid Hormone; Ado: Adenosine; ATP: Adenosine-Triphosphate; ADA: Adenosine-Deaminase; MAO: Monoamine Oxidase; P1R: Purinerg Receptor type 1; P2R: Purinerg Receptor type 2.
ACTH: Adrenocorticoid Hormone; Ado: Adenosine; ATP: Adenosine-Triphosphate; ADA: Adenosine-Deaminase; MAO: Monoamine Oxidase; P1R: Purinerg Receptor type 1; P2R: Purinerg Receptor type 2.
Model 2: How does dexamethasone affect the plasma levels of various hormones, ATP and adenosine, in stressful situations?>
The various types of synthetic glucocorticoids are used for anti-inflammatory and immunosuppressant therapies with the known risks of strong negative side effects, like osteoporosis. In this work, DXM was chosen as the “model molecule” to demonstrate the mechanism of the acceleration of osteoporosis initiated by the lack of mineralocorticoid effects leading to pathologic alterations in the plasma levels of various hormones. These extra and intracellular processes are synthesized in Model 2 (Figure 2). The dominating effect of DXM is the blocking of ACTH production, resulting in the ceasing of cortisol and aldosterone production in the cortex of the adrenal gland. In this dangerous situation, the urinary loss of Na+ and the plasma level of K+ are increased. However, the potassium ions exert direct stimulatory effects on aldosterone secretion, which is only partly independent of the renin-angiotensin system. A small increase in plasma K+ levels already elicits a rise in plasma aldosterone levels [14]. The life-saving reaction is that the increased amounts of plasma K+ trigger the aldosterone production in the adrenal gland, but in an uncontrolled elevated form, as the aldosterone synthesis will later be augmented by the activation of renin-angiotensin II system [14]. High levels of aldosterone induce high production of PTH [12], accelerating the resorption of bones [8]. This osteoporotic trend is further strengthened by DXM which increases the urinary and intestinal loss of Ca2+ [3]. It can be apparent that the altered processes of mineral ion metabolism induced by DXM and other direct glucocorticoid effects can be the main factors of osteoporosis and bone fractures. In parallel, DXM elevates the adrenaline production by the medulla of the adrenal gland (even better than cortisol could be) [15], causing high extracellular levels of ATP and Ado. Simultaneously, DXM enhances the activity of MAO, degrading the increased amounts of adrenaline [16]. It can also be concluded that these changes caused by DXM and other GS molecules represent a special form of stress response, which can be called a “GS-induced stress situation”. The blockade of ACTH production in the pituitary gland, when even great amounts of extracellular Ado molecules are ineffective, is also an additional part of dysregulations. The other main factor is the lack of mineralocorticoid activity in the GS medicaments, leading to a series of pathologic states such as high K+ levels, uncontrolled increase in aldosterone and PTH synthesis, and finally, secondary hyperparathyroidism, which induces accelerated osteoporosis and frequent bone fractures. All these negative factors together can still cause complex disturbances in the regulation of stress responses during steroid therapy.
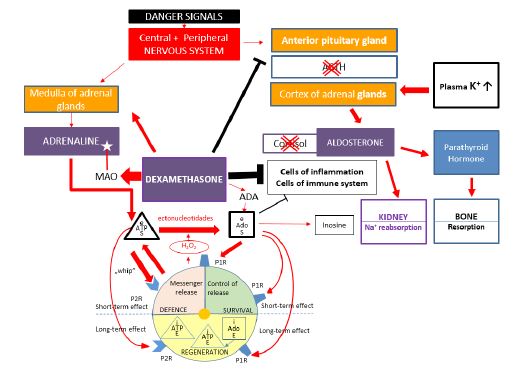
Figure 2: Effects of dexamethasone on the plasma levels of various hormones, ATP and adenosine in stressful situations.
ACTH: Adrenocorticoid Hormone; Ado: Adenosine; ATP: Adenosine-Triphosphate; ADA: Adenosine-Deaminase; MAO: Monoamine Oxidase; P1R: Purinerg Receptor type 1; P2R: Purinerg Receptor Type 2
ACTH: Adrenocorticoid Hormone; Ado: Adenosine; ATP: Adenosine-Triphosphate; ADA: Adenosine-Deaminase; MAO: Monoamine Oxidase; P1R: Purinerg Receptor type 1; P2R: Purinerg Receptor Type 2
Model 3: The synthesized model of extracellular changes in the amounts of mineral ions and hormones leading to osteoporosis caused by dexamethasone therapy
In Model 3 (Figure 3) are demonstrated those main extracellular changes in the mineral ion metabolism and hormone levels which play the main roles in the induction of accelerated osteoporosis caused by DXM. Accepting and using the beneficial effects of elevated glucocorticoid activity of synthetic steroids in the anti-inflammatory and immunosuppressant therapies, we emphasize now that in their negative side effects causing osteoporosis, the lack of their mineralocorticoid activity plays a crucial role. The main parts of pathologic changes can be attributed to this fact, including the characteristic features of osteoporosis, the resorption of bones and the loss of Ca2+ [3]. DXM can achieve such conditions: a) by strongly blocking ACTH production in the pituitary gland, leading to the stop of cortisol and aldosterone synthesis in the adrenal glands. This is a transient but critical state because of the total lack of urinary Na+ reabsorption and a toxic elevation of plasma K+ levels which can come into being. b) However, at a certain high level of K+, the production of aldosterone by the cortex of adrenal glands can be restarted but in a large amount and uncontrolled form. c) High aldosterone levels will involve a great increase in the production of PTH. This hormone has a central role. It regulates both bone production and Ca2+ metabolism. d) DXM can stimulate both PTH production and the intestinal and urinary loss of Ca2+ e) Furthermore, DXM can inhibit the production of new bone tissue and sex hormones, which prevent bone resorption [3].
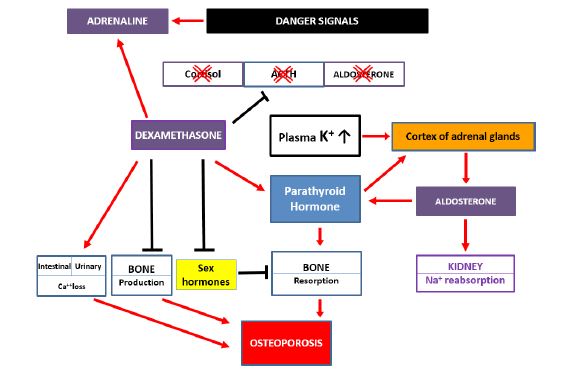
Figure 3: The synthesized model of extracellular changes in the amounts of mineral ions and hormones leading to osteoporosis caused by dexamethasone therapy.
Conclusion
In this review, there are two novelties. This is the first synthesis of those processes which show the qualitative and quantitative intracellular and extracellular changes in the molecules of the “extracellular ATP-adenosine danger cycle” and the plasma levels of main stress hormones during a “natural stress response” extended by the role of parathyroid hormone. Furthermore, it is pointed out that the absence of mineralocorticoid activity in the synthetic glucocorticoids leading to the high elevation of plasma K+ levels is one of the main triggers of complex disturbances leading to secondary hyperparathyroidism, osteoporosis and frequent bone fractures.
The presented new models can contribute to a better understanding of the intra - and extracellular processes in natural stress responses and the molecular mechanisms of accelerated severe osteoporosis induced by synthetic glucocorticoids used as basic drugs in anti-inflammatory and immunosuppressant therapies. Besides, it is suggested that “GIOP can be a special, drug-induced form of stress responses.” In this work, DXM was used as a “model drug” because it owned the most extreme glucocorticoid and mineralocorticoid features of synthetic glucocorticoids. In the actions of other members of this drug family, used more frequently in practice, the same tendencies are present, but to a less extent. However, in pulse therapy or during long lasting treatments, the lack of mineralocorticoid effects of these medicaments will also involve the elevation of plasma K+ levels leading to seconder hyperparathyroidism and accelerated osteoporosis according to the extent of disturbance caused. The simultaneous demonstration of intra- and extracellular processes during stress responses and GS therapy can elucidate the sensitive, tight, vital and signal-dependent linkage between them: the greater stress hormone signals, the stronger responses by the involvement of mineral ions and the molecules of ATP-adenosine cycle. Finally, this composition of earlier observations from a new approach due to the increased plasma levels of K+ can open a new direction in the therapy of GIOP [17,18].
Author Statements
Data availability statement: This review is based on the articles presented in the chapter References.
Acknowledgement: The authors thank Anikó Pasztenak the drafting of figures.
Author’s contributions: S.S. gave the concept and wrote the article; A.N. helped the work both intellectually and technically; J SZ, SS Jr. and ZS B. took part in the discussion of the concept of work, TT gave the clinical background.
Conflict of interest: The authors disclose any conflicts among them.
Funding: The authors declare that no funds or other supports were received for the manuscript.
Ethical standards: The authors followed all the substantial ethical standards during the study period.
Consent for publication: Consent for publication was obtained from all authors.
References
- NIH Consensus Development Panel on Osteoporosis: Prevention, Diagnosis, and Therapy. Osteoporosis prevention and therapy. JAMA. 2011; 285: 785-95.
- Föger-Samwald U, Dovjak P, Azizi-Semrad U, Kerschan-Schindl K, Pietschmann P. Osteoporosis: pathophysiology and therapeutic options. EXCLI Journal. 2020; 19: 1017-1037.
- Canalis A, Mazziotti A, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporosis International. 2007; 1319-1328.
- Kazakou P, Nicolaides NC, Chrousos GP. Basic concepts and hormonal regulators of the stress system. Hormone Research in Paediatrics. 2023; 96; 8-16.
- Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria are key components of the stress response. TRENDS in Endocrinology and Metabolism. 2007; 18: 190-198.
- Sipka S, Pázmándi K, Sipka S JR, Bruckner G, Bodnár Zs. Quantitative and qualitative changes in the Ca2+, ATP-adenosine cycle and ROS metabolisms depending on the strength of danger signals during apoptosis and necrosis compared to homeostasis in various danger/stress situations. Hematology and Oncology: Current Research. 2022; 5: 1019.
- Benedek TG. History of the development of corticosteroid therapy. Clin Exp Rheumatol. 2011; 5: 5-2.
- Adler RA, Hochberg MC. Suggested guidelines for evaluating and treating glucocorticoid-induced osteoporosis for the Department of Veterans Affairs. Archives of International Medicine. 2003; 163: 2619-2624.
- Szabó J, Kósa E, Tóth IE, Bruckner G. Effect of adenosine and its metabolites on the hypothalamic-pituitary-adrenal axis. Nutritional Biochemistry. 1995; 6: 334-339.
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: contemporary view with implications for physiology and medicine. Pharacol Rev. 2004; 56: 331-349.
- Kytrib-Zajac B, Kawecka A, Nasadiuk K, Braczko A, Stawarska K, Caiazzo E, et al. Drugs targeting adenosine signalling pathways: a current view. Biomedicine and Pharmacotherapy. 2023; 165: 115184.
- Stanic BM, Ilincic B, Zeravica R, Ivanovski DM, Cabarkapa V, Milovic R. The importance of the correlation between aldosterone and parathyroid hormone in patients with primary hyperparathyroidism. Int J Endocrinol. 2022: 3804899.
- Yaribeygi H, Maleki M, Butler AE, Jamailahmadi T, Sahebkar A. Molecular mechanisms linking stress and insulin resistance. EXCLI Journal. 2022; 21: 317-334.
- Bhagavan NV, Chung-Eun-Ha. Regulation of corticosteroid secretion, in Chapter 30. Essential of Medical Biochemistry (Second Edition). 2015: 559-575.
- Sharara RI, Joachim M, Pacak K, Majzoub A. Glucocorticoid treatment –effect on adrenal medullary catecholamine production. Shock. 2010; 33: 213-217.
- Ou XM, Chen K, Shih JC. Glucocorticoid and androgen activation of monoamine oxidase A is regulated differently by R1 and Sp1. J Biol Chem. 2006; 281:21512-21525.
- Humphrey MB, Russell L, Danila MI, Fink HA, Guyatt G, Cannon M, et al. 2022 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-induced Osteoporosis. Arthritis Rheumatol. 2023; 75:2088-2102.
- Chiodini I, Merlotti D, Falchetti A, Gennari L. Treatment options for glucocorticoid-induced osteoporosis. Expert Opinion on Pharmacotherapy. 2020; 21:721-732.
