
Open Access, Volume 10
Selexipag as third add-on therapy in a thalassemic patient with pulmonary hypertension
Lucia Tricarico1; Michele Correale2*; Ester Maria Lucia Bevere1; Antonio Ruggiero3; Erminia Guerriero4; Ludovica Trupo4; Maria Grazia Roberti5; Filomena Sportelli5; Natale Daniele Brunetti1
1Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy.
2Cardiothoracic Department, Policlinico Riuniti University Hospital, Foggia, Italy.
3Policlinico Riuniti University Hospital, Foggia, Italy.
4Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy.
5Transfusional Medicine Unit, Policlinico Riuniti University Hospital, Foggia, Italy.
Michele Correale
Cardiothoracic Department, Policlinico Riuniti University Hospital, Foggia, Italy.
Tel: +39 3331320864 & +39 0881 745424;
Email: michele.correale@libero.it
Received : November 17, 2024,
Accepted : December 12, 2024
Published : December 16, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Pulmonary Hypertension (PH) is a frequent clinical complication in patients with thalassemia major. PH is caused by increased pulmonary resistance induced by chronic hypoxia, iron-phosphatidylserine-expressing hematologic debris, free hemoglobin, and other circulating angiotrophic factors. Chronic transfusion therapy and concomitant chelation therapy have been shown to prevent PH, whereas splenectomy was associated with a higher risk of pulmonary embolism and PH. Patients affected by β-thalassemia with PH are usually included among PH patients with unclear and/or multifactorial mechanisms (group 5), however, such patients often show others comorbidities (as pulmonary thromboembolic disease). Therapeutic options in these patients are not well defined, even in the absence of data from randomized studies. We report the case of a 50-year-old man affected by thalassemia major, diabetes, chronic HCV-hepatitis, osteoporosis, hypogonadotropic hypogonadism, with a prior episode of septic embolism and consequent inoperable post-thromboembolic PH, undergoing sequential combination therapy for PAH (sildenafil plus macitentan) plus selexipag, with very long follow-up. Sequential combination therapy for PAH (sildenafil plus macitentan) plus selexipag, in thalassemic patient with PH, may be safe and apparently rapidly effective in reducing dyspnea and improving performance.
Keywords: Pulmonary arterial hypertension; Thalassemia; Macitentan; Selexipag; Chronic thromboembolic pulmonary hypertension.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Correale M (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Tricarico L, Correale M, Lucia Bevere EM, Ruggiero A, Guerriero E, Trupo L, Grazia Roberti M, et al. Selexipag as third add-on therapy in a thalassemic patient with pulmonary hypertension. Open J Clin Med Case Rep. 2024; 2309.
Background
Pulmonary Hypertension (PH) is a frequent clinical complication in patients with thalassemia major. PH is caused by increased pulmonary resistance induced by chronic hypoxia, iron-phosphatidylserine-expressing hematologic debris, free hemoglobin, and other circulating angiotrophic factors. Chronic transfusion therapy and concomitant chelation therapy have been shown to prevent PH, whereas splenectomy was associated with a higher risk of pulmonary embolism and PH. Patients affected by β-thalassemia with PH are usually included among PH patients with unclear and/or multifactorial mechanisms (group 5), however, such patients often show other comorbidities (as pulmonary thromboembolic disease). Therapeutic options in these patients are not well defined, even in the absence of definitive data from randomized studies [1].
Case Report
We report the case of a 50-year-old man affected by thalassemia major (transfusion dependent), diabetes, chronic HCV-hepatitis, osteoporosis, hypogonadotropic hypogonadism, with previous splenectomy and a prior episode of septic pulmonary embolism in 2007 and consequent inoperable post-thromboembolic PH, that was referred for worsening dyspnea (WHO functional class III) and palpitations. Therapy was started (digitalis, diuretics and anticoagulants), and, since 2010 for the increasing of dyspnea (WHO functional class IV) sildenafil 20 mg TD was added; clinical condition remained stable for a long period (WHO functional class II). In 2015 he complained of worsening dyspnea (WHO functional class III) and since November 2015 started macitentan 10 mg die with an early improvement of dyspnea (WHO functional class of II). By combination therapy clinical condition (WHO functional class II) remained stable several years (WHO functional class II), without any adverse events (Figure 1).
In January 2022, dyspnea increased and echocardiographic examination showed enlarged right cardiac chambers, severe tricuspid regurgitation, and increased pulmonary arterial systolic pressure (PAsP 92 mmHg). Six-minute walking test showed an impaired functional capacity (165 m). Selexipag 200 mcg BID (later increased to 600 mcg bid) was added, with an observed early improvement of clinical condition and dyspnea (WHO functional class of II). After 12 months of selexipag, 6MWT performance also improved to 235 m and there was a reduction of the echocardiographic value of PAsP to 62 mmHg and of the degree of tricuspid regurgitation to moderate (Figure 2). By October 2024, the patient continues to be classified in WHO functional class II and remains on triple-specific PAH therapy (Sildenafil, macitentan, and selexipag). Throughout this time, the patient has been stable, with no hospitalizations, and the treatment has been well-tolerated without significant side effects.

Figure 1: Follow up and therapy timeline.
WHO FC: World Health Organization functional class.
WHO FC: World Health Organization functional class.
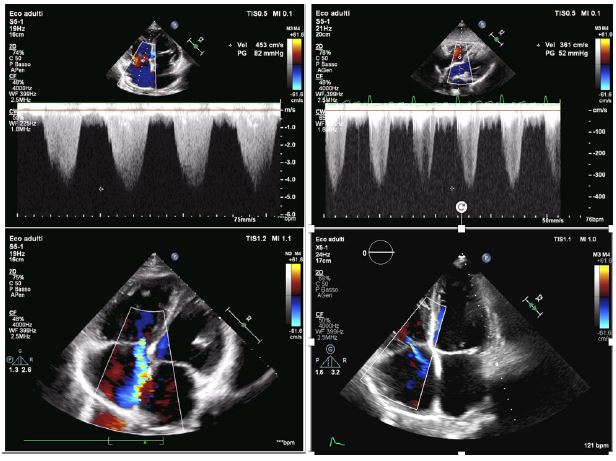
Figure 2: Echocardiographic features before (A,C) and after (B,D) adding selexipag to dual therapy. A, B show tricuspid regurgitation peak gradient before (A) and after (B) adding selexipag to dual therapy; C, D the degree of tricuspid regurgitation before (C) and after (D) adding selexipag to dual therapy.
Vel: Velocity; PG: Pressure Gradient.
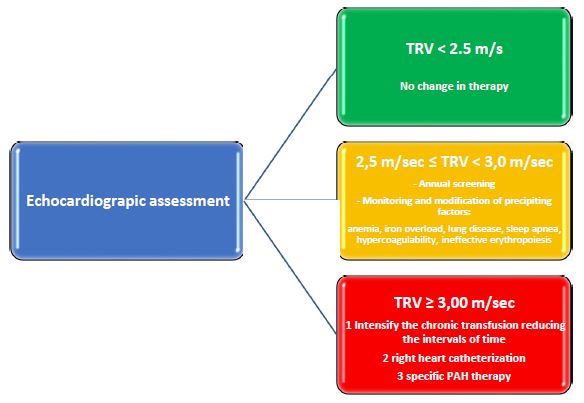
Figure 3: Management algorithm based on echocardiographic assessment in thalassemia patients.
TRV: Tricuspid Regurgitation Jet Velocity; PAH: Pulmonary Arterial Hypertension.
TRV: Tricuspid Regurgitation Jet Velocity; PAH: Pulmonary Arterial Hypertension.
Discussion
Patients with β-thalassemia with PH are usually included among PH cases with unclear and/or multifactorial mechanisms (group 5); however, such patients often show other comorbidities (as pulmonary thrombo-embolic disease). Chronic hypoxia and the resultant high cardiac output state contribute to mild increase in pulmonary artery pressure. However significant PH is caused by an increase in pulmonary vascular resistance induced by chronic hypoxia, iron-phosphatidylserine-expressing hematologic debris, free hemoglobin, and other circulating angiotrophic factors [2].
Echocardiography could be an important tool to assess response to therapy. Recently, the WebThal study [3], considering that PAsP values determined by Tricuspid Regurgitation Jet Velocity (TRV) associated with survival, has provided a management algorithm that can be useful to determine treatment escalation in thalassemia patients with PAH (Figure 3).
Chronic transfusions and concomitant chelation therapy have been shown to prevent PH in both forms of thalassemia, as shown in the OPTIMAL CARE study, while splenectomy was associated with a higher risk of pulmonary embolism and PH [4].
PH specific therapies have been only assessed in small series or case reports. Since targeted PH therapy have no proven benefit, their use should be restricted to patients with clear hemodynamic evidence of pre-capillary PH [5].
Although there are insufficient data for general recommendations, some small studies have shown potential benefits of targeted PH therapies in β-thalassemia patients [6]. Sildenafil has been showed to improve NYHA functional class, echocardiographic evidence of pulmonary hypertension and 6MWT [7-9]. A few studies also demonstrated a role for endothelin receptor antagonist [10-12] or epoprostenol [13] in such patients.
Therapeutic options in group 5 patients are not well defined and not supported by randomized clinical studies. According to ESC/ERS guidelines recommendations, these patients need careful assessment; therapy should be tailored according to underlying conditions.
In our case, transfusion therapy failed, so selexipag administration was started in order to delay disease progression and reduce risk of hospitalization.
To the best of our knowledge, no previous reports are available on the administration of selexipag in thalassemic patient with PH without adverse events after almost two years and 9 months of therapy.
In conclusion, selexipag, in thalassemic patient with PH, may represent a safe option and apparently rapidly effective in reducing dyspnea, TRV values and improving performance at 6MWT. Further large and multicenter studies are warranted to confirm such preliminary data.
Keypoints
Patients affected by β-thalassemia with PH are usually included among PH patients with unclear and/or multifactorial mechanisms (group 5). Therapeutic options in these patients are not well defined. Sequential combination therapy for PAH (sildenafil plus macitentan) plus selexipag, in this thalassemic patient with CPETH, may be safe and apparently rapidly effective in reducing dyspnea and improving performance.
References
- Sasiprapha T, Pussadhamma B, Sibmooh N, Sriwantana T, Pienvichit P, et al. Efficacy and safety of inhaled nitrite in addition to sildenafil in thalassemia patients with pulmonary hypertension: A 12-week randomized, double-blind placebo-controlled clinical trial. Nitric Oxide. 2022; 120: 38-43.
- Paul A, Thomson VS, Refat M, Al-Rawahi B, Taher A, et al. Cardiac involvement in beta-thalassaemia: Current treatment strategies. Postgrad Med. 2019; 131(4): 261-267
- Longo F, Corrieri P, Origa R, Barella S, Sanna PMG, et al. Changing patterns of thalassaemia in Italy: A WebThal perspective. Blood Transfus. 2021; 19(3): 261-268.
- Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The Optimal Care study. Blood. 2010; 115(10): 1886-92.
- Anthi A, Orfanos SE, Armaganidis A. Pulmonary hypertension in β thalassaemia. Lancet Respir Med. 2013; 1(6): 488-96.
- Pinto VM, Musallam KM, Derchi G, Graziadei G, Giuditta M, et al. Webthal project. Mortality in β-thalassemia patients with confirmed pulmonary arterial hypertension on right heart catheterization. Blood. 2022; 139(13): 2080-2083.
- Morris CR, Kim HY, Wood J, Porter JB, Klings ES, et al. Thalassemia Clinical Research Network. Sildenafil therapy in thalassemia patients with Doppler-defined risk of pulmonary hypertension. Haematologica. 2013; 98(9): 1359-67.
- Derchi G, Balocco M, Bina P, Caruso V, D’Ascola DG, et al. Efficacy and safety of sildenafil for the treatment of severe pulmonary hypertension in patients with hemoglobinopathies: Results from a long-term follow up. Haematologica. 2014; 99(2): e17-8.
- Correale M, De Rosa F, Ieva R, Di Biase M, Brunetti ND. Long-term treatment with high-dose of sildenafil in a thalassemic patient with pulmonary hypertension. Monaldi Arch Chest Dis. 2012; 78(2): 105-6.
- Anthi A, Tsangaris I, Hamodraka ES, Lekakis J, Armaganidis A, et al. Treatment with bosentan in a patient with thalassemia intermedia and pulmonary arterial hypertension. Blood. 2012; 120(7): 1531-2.
- Correale M, Zicchino S, Monaco I, Brunetti ND, Di Biase M. Association therapy with macitentan added to sildenafil in a thalassemic patient with pulmonary hypertension. Int J Cardiol. 2016; 220: 80-1.
- Takagi K, Kasai H, Tani H, Sakao S, Sugiura T, et al. Macitentan Administration for Pulmonary Hypertension Due to β-thalassemia with Multiple Organ Failure: A Case Report. Intern Med. 2023. doi: 10.2169/internalmedicine.2307-23.
- Ussavarungsi K, Burger CD. Pulmonary arterial hypertension in a patient with β-thalassemia intermedia and reversal with infusion epoprostenol then transition to oral calcium channel blocker therapy: Review of literature. Pulm Circ. 2014; 4(3): 520-6.
