
Open Access, Volume 10
Congenital cataract with global developmental delay and microcephaly in three Tunisian families: A new recessive syndrome?
Chograni M1; Ouerteni Ines2; Kraoua Lilia2
1Laboratory of Human Genetics, Faculty of Medicine of Tunis, University Tunis El Manar, Tunis, Tunisia.
2Congenital and Hereditary Disorders Department, Charles Nicolle hospital, Tunis, Tunisia.
Manèl Chograni
Laboratory of Human Genetics, Faculty of Medicine of Tunis, University Tunis El Manar, 17 avenue Jebel Lakhdar, Tunis 1007, Tunisia.
Email: chogranimanel@yahoo.fr
Received : October 04, 2024,
Accepted : October 28, 2024
Published : October 31, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Congenital cataract is one of the most genetically heterogeneous ocular malformations causing visual impairment and blindness in children. The present work aimed to evaluate the characteristics of extraocular manifestations in Tunisian patients with Autosomal Recessive Congenital Cataract. In this cross-sectional study, seven patients belonging to three unrelated Tunisian families, and whose parents were first cousins, were enrolled in the study. Data regarding weight, cataract duration and subtype, age at diagnosis, microcephaly, Global Developmental Delay (GDD), and psychomotor development profile were collected, and a full ophthalmic examination was conducted. We report seven affected patients with Autosomal Recessive Congenital Cataract in association with GDD and microcephaly syndrome. The affected patients had congenital cataract, GDD and microcephaly suspected since birth with no facial dysmorphism. The reported association is not due neither to a metabolic disease seeing the normal investigations in all affected patients had congenital microcephaly, nor to chromosomal abnormalities. Review of published reports and the use of the London Dysmorphology Database suggest that the three families present a new syndrome never described to date. We observed significant associations between autosomal recessive congenital cataract, GDD and microcephaly in the three families. Our findings suggest a new familial syndrome that we are working to identify the genetic cause leading to such syndrome in children, and can provide reference and ideas for underlying these defects and treatment plans in the future.
Keywords: Congenital cataract; Microcephaly; Global developmental delay; Association; New syndrome.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Chograni M (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Chograni M, Ines O, Lilia K. Congenital cataract with global developmental delay and microcephaly in three Tunisian families: A new recessive Syndrome?. Open J Clin Med Case Rep. 2024; 2299.
Introduction
Cataract can be defined as opacity of the crystalline lens [1]. It has also been reported as the commonest congenital ocular malformation, with a prevalence of 2.7 per 10 000 births [2]. Bilateral congenital cataract is the most common cause of treatable childhood blindness, accounting for 5% to 2% of blindness in children worldwide [3]. Different clinical aspects can be observed: from cataract with ocular and/or systemic anomalies to polymalformative syndrome, skeletal, dermatological, neurological, metabolic, and genetic or chromosomal diseases. Non-syndromic global developmental delay is an intellectual disability which severity is determined by the IQ and with no other consistent features than global developmental delay. Microcephaly is a congenital disorder in which the head circumference is greater than two Standard Deviations (SD) below the age and sex-related population mean [4].
The aim of this study was to review a group of seven patients from three Tunisian families with a phenotype associating congenital cataract, Global Developmental Delay (GDD) and microcephaly.
Material and methods
Seven patients belonging to three unrelated Tunisian families (F1, F2, and F3) were screened at the Congenital and Hereditary Disorders Department at Charles Nicolle Hospital (Tunis, Tunisia) because of an unreported association between congenital cataract, GDD and microcephaly. All patients were born to healthy consanguineous parents; F=1/16 (Figure 1). They presented with bilateral posterior polar cataract clinically suspected at birth. Significant physical disability became apparent for all patients by the age of 15 to 18 months when they failed to walk. They also had significant delay in speech development. In fact, the seven affected patients were developmentally delayed with moderate GDD and no dysmorphic features. Informed consent was obtained from participants after the protocol was previously approved by the Institutional Review Board of the Congenital and Hereditary Disorders Department at Charles Nicolle Hospital (Tunis, Tunisia), and we confirm that the whole study protocol was approved by the named IRB, and all methods were performed in accordance with the relevant guidelines and regulations.

Figure 1: Pedigrees of the three studied families: F1, F2 and F3 showing autosomal recessive inheritance of the congenital cataract. The asterisk indicates not examined.
Family 1 (F1): This family is originally from the northwestern of Tunisia. She presented with three affected patients; the older one is the girl (IV35) referred to our department because of congenital cataract associated to global developmental delay and microcephaly and whose affected brothers were characterized also with an association between congenital cataract, global developmental delay and a lower limit of head Circumference (CP).
Family 2 (F2): This family is originated from northern Tunisia. She had two affected patients with congenital cataract, global developmental delay and microcephaly. We noted that their father died after a traumatic accident and their grand-mothers (II5 and II7) had late-onset cataract.
Family 3 (F3): This family came from the southeast of Tunisia. She was referred to the Congenital and Hereditary Disorders department because of two affected patients with congenital cataract, global developmental delay.
Results
Origins and pedigrees
We assumed that all families are of Arab ethnicity, originated from Tunisia. The three families were unrelated but all were consanguineous; the degree of relationship was first cousins. We considered that the designed phenotype: congenital cataract, global developmental delay with microcephaly was concordant with autosomal recessive inheritance
Patient 1 (F1: IV35)
This girl aged 9 years was examined for the first time in our department on account of an association between congenital cataract, and global developmental delay. She presented a normal development to thrive (weight and height). Ophthalmologic examinations confirmed bilateral cataract detected by the age of 6 years, suspected since birth and extracted at 7 years for both eyes. The cataract was of the posterior polar type and was not associated to any other ocular feature. The girl was developmentally delayed; she was labeled by a psychomotor retardation, in fact she started to walk by the age of 2 years and pronounced the first word at 5 years.
In addition, she presented a difficulty to shake hands, close eyes and stuttering. Head circumference measurement revealed congenital microcephaly with -2.8 SD with no facial dysmorphism. She had no convulsion and neurological examination was normal. Investigations showed normal visual function, normal karyotype 46, XX (600-bands resolution), normal metabolic screening including normal Fehling reaction and normal thin layer chromatography of reducing sugars, normal plasmatic amino acid and urine organic chromatography. Magnetic resonance imaging (MRI) of the brain and muscle electromyography were normal.
Patient 2 (F1: IV36)
The propositus brother, aged 5 years, had normal thrive development. Ocular exploration at 3 years showed bilateral cataract which was suspected since birth. The cataract was of the posterior polar type and was isolated. He had developmental delay and motor retardation; he walked at age 2 years and spoke at age 5 years.
He was examined again at the age of 15 years, he spoke with a slurred speech, he had no autonomy and could not dress alone. He presented a lower limit of CP and had no facial dysmorphism (Figure 2A). No convulsion was assigned, and neurological examination was normal. Investigations showed normal visual function, normal karyotype 46, XY (600-bands resolution), normal metabolic screening including normal Fehling reaction and normal thin layer chromatography of reducing sugars, normal plasmatic amino acid and urine organic chromatography. Magnetic resonance imaging of the brain and muscle electromyography were normal.
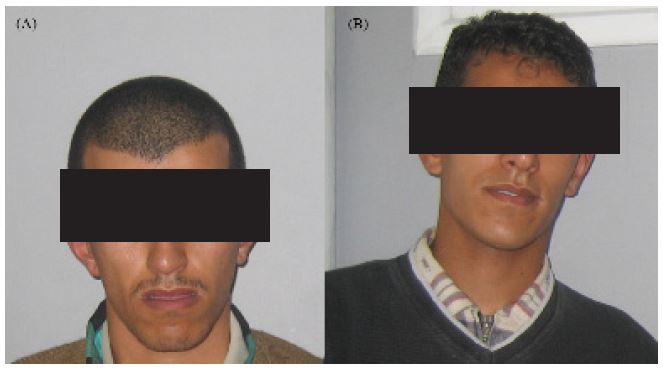
Figure 2: Facial appearance in the two patients IV36 (A) and IV37 (B) from family 1 showing no dysmorphic features.
Patient 3 (F1: IV37)
The propositus second brother was examined for the first time in our department at the age of 12 years because of congenital cataract and global developmental delay. He presented normal weight and height.
Ophthalmologic examinations at 4-year age revealed bilateral cataract of posterior polar type and suspected since birth. He had undergone cataract extraction for both eyes at the age of 5 years. Diagnosis of cataract evinced that it is not associated with another ocular anomaly. The boy showed a developmental delay with psychomotor retardation, in fact he spoke with a stutter and he had difficulty to learn. He had difficulties to shake hands, too. Head circumference measurement showed a lower limit of CP. He had no facial dysmorphism (Figure 2B), no convulsion and neurological examination was normal. Investigations showed normal visual function and normal biological tests (karyotype, metabolic screening). Magnetic resonance imaging of the brain and muscle electromyography were normal.
Patient 1 is a female, aged 28 years, was examined for the first time in our department at 4 years because of congenital cataract associated to global developmental delay. She had normal development to thrive.
Ophthalmologic examinations revealed unilateral cataract suspected since birth which became apparent at 3 years. She had undergone an extracapsular extraction and implantation of the posterior chamber at the age of 5 years.
The cataract was of the posterior polar type and was not associated to any other ocular anomaly. The girl had developmental delay; she could not learn nor write. She showed also psychomotor retardation with a significant delay in speech development and a slurred speech. She had severe microcephaly at -5.5 SD with no facial dysmorphism (Figure 3A). She never had convulsion but neurological examination at age 21 years showed mild axial hypotonia and mild spastic walk. Investigations showed normal visual function, normal karyotype 46, XX (600-bands resolution), normal metabolic screening including normal Fehling reaction and normal thin layer chromatography of reducing sugars, normal plasmatic amino acid and urine organic chromatography. Magnetic resonance imaging of the brain and muscle electromyography were normal.
Patient 5 (F2: IV16)
This younger sister, aged 17 years, were examined in our department by age 6 years. She had normal weight and height. Ophthalmologic examination showed bilateral cataract detected at 6 years and suspected since birth. She had undergone an extracapsular extraction and implantation of the posterior chamber at the age of 6 years for both eyes. Cataract was of the posterior polar type, associated with white dots in the anterior vitreous, strabismus of the left eye and retinal dystrophy. She had developmental delay with learning and reading disabilities. She presented also a psychomotor retardation with a significant delay in speech, and she speaks with a stutter. She had severe microcephaly with -7.6 SD with no dysmorphic features (Figure 3B). She was examined again at age of 13 years; no convulsion was detected, and neurological examination showed mild axial hypotonia, increased tone and contracture in the lower limbs with flexed knees, tetrapyramidal syndrome. Biological investigations showed a trouble in visual function marked by a decrease in visual acuity, normal karyotype 46, XX (600-bands resolution), normal metabolic screening including normal Fehling reaction and normal thin layer chromatography of reducing sugars, normal plasmatic aminoacid and urine organic chromatography. Muscle electromyography was normal and magnetic resonance imaging of the brain performed at 13 years showed a small ischemic parietal lesion.
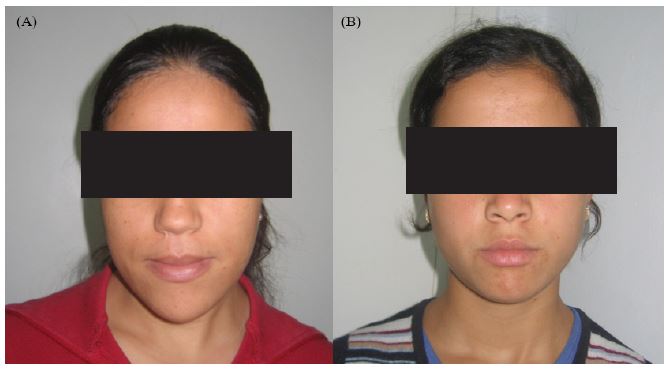
Figure 3: Facial appearance in the two patients IV13 (A) and IV16 (B) from family 2 underlining the absence of facial dysmorphism.
Patient 6 (F3: IV11)
This female patient was referred to our department at 12 years. She had normal statural development. At age 31/2 years, exploration of the fundus revealed bilateral cataract detected and extracted since birth. Cataract was of the posterior polar type and was associated to an alteration of the pigment epithelium, leucorrhea was also observed. The girl had developmental delay and psychomotor retardation; she walked and spoke at 2 years and while speaking she confused the letters. She had microcephaly at -3.8 SD, she had no facial dysmorphism, no convulsion and neurological examination was normal. Ophthalmologic investigations showed a trouble in visual function marked by night-blindness and decrease in visual acuity. Biological investigations including karyotyping with R-bands, 46, XX (600-bands resolution), metabolic screening including Fehling reaction and thin layer chromatography of reducing sugars, plasmatic amino acid and urine organic chromatography were normal. She had infantile genitalia and no signs of puberty at 12 years. Magnetic resonance imaging of the brain and muscle electromyography were normal.
Patient 7 (F3: IV12)
The brother was 5 years old when referred to the department that was diagnosed. He had normal weight and height. At 3 years ophthalmologic examination showed unilateral cataract few days after birth. Cataract was of the posterior polar type and not associated to any other ocular features. The patient had subnormal development, in fact he walked at 1 year and a half, spoke at 2 years and at 5 years he had a speech impediment. He had microcephaly with -5.6 SD. He showed no dysmorphic features, no convulsion and neurologic examination was normal. Ophthalmologic investigations showed normal visual function. Biological investigations normal karyotype 46, XY (600-bands resolution), normal metabolic screening including normal Fehling reaction and normal thin layer chromatography of reducing sugars, normal plasmatic amino acid and urine organic chromatography and normal menarche. Magnetic resonance imaging of the brain and muscle electromyography were normal.
Congenital cataract associated to GDD and microcephaly
All affected individuals showed a delayed motor and an intellectual development associated to congenital cataract and microcephaly. Congenital cataract was the first recognized feature in all 7 patients. It was revealed either isolated or associated to other ocular anomalies.
Discussion
The purpose of this study was to review familial cases presenting congenital cataract associated to GDD and microcephaly with no dysmorphic features. To our knowledge there is no alikehood of an underlying known syndrome in this group.
Here we report three Tunisian consanguineous families with seven affected patients. There was no failure to thrive for all patients. They had congenital cataract of the posterior polar type which was bilateral for 5 patients and unilateral in 2 cases. The cataract found to be isolated in 5 patients and associated to other ocular features in two of them: white dots in the anterior vitreous, strabismus of the left eye and retinal dystrophy with decrease in visual acuity in patient IV16 from F2 and alteration of the pigment epithelium and leucorea associated to a decrease in visual acuity and night-blindness in patient IV11 from F3. The seven patients presented developmental and psychomotor delay with mild to moderate global developmental delay, in fact all of them had walked and spoken at late age and had behavior and schooling difficulties. Five patients had microcephaly suspected since birth and two had a lower limit of head circumference (-1 SD). No convulsion had been assigned in all patients and neurologic examination was normal except the two sisters belonging to F2 who showed mild axial hypotonia and a mild spastic walk (IV13) and mild axial hypotonia, increased tone and contracture in the lower limbs with flexed knees, tetrapyramidal syndrome (IV16). All patients showed normal investigations, except the patient IV16 (F2) whose MRI revealed a small ischemic parietal lesion. In fact, embryologically the lens is the first component of the eye that begins to be differentiated and it is to the sixth week of embryonic life followed by retinal and corneal differentiations. In fact, the reports of the fetal vascularization with the lens are of great importance in the genesis of some anomalies associated with some forms of cataracts; by the third week after fertilization appears the first vascularization as a hyaloid artery that held the slot fetal located on the underside of the blank eye. This artery reaches the posterior pole of the future lens in one week, expands on its face and becomes the posterior vascular tunic of the lens that surrounds it closely. During the last months, all vessels must be disappeared by gradual regression from back to front. But in some abnormal cases the remnants of the hyaloid artery persisted leading to the installation of the posterior polar capsular cataract which is a minor form of congenital cataract. In addition, partial persistence of fetal vascular system can cause all sorts of abnormalities in the vitreous or anterior chamber that may be associated with the presence of congenital cataract. However, for strabismus it is a consequence of malvision rather than an ocular abnormality associated to cataract.
We highlighted that the reported association between congenital cataract, global developmental delay and microcephaly is not due neither to a metabolic disease seeing the normal investigations in all affected patients had congenital microcephaly, nor to chromosomal abnormalities. Moreover, all reported syndromes presenting such an association are characterized by other cardinal signs in that ARCC found to be associated with microcephaly in a typical methylmalonic aciduria [5] with no sign of GDD but in association with encephalopathy, dystonia and spasticity. Martsolf syndrome is also associated with cataracts, short stature, hypogonadism and severe GDD. It appears to be autosomal recessive [6]. Micro syndrome was first reported by Warburg et al [7] as an autosomal recessive condition characterised by microcornea, cataract, optic atrophy, microcephaly, severe GDD, hypotonia, spastic diplegia, cortical gyral abnormalities, hypoplasia of corpus callosum, and hypothalamic hypogenitalism. Marinesco-Sjogren syndrome is an autosomal recessive multisystem disorder characterised by cataracts, cerebellar hypoplasia, growth retardation, mild-moderate GDD, hypotonia, demyelinating neuropathy, seizures, hypogonadism, and skeletal abnormalities [8]. Early-onset Cockayne Syndrome (CS2) is an autosomal recessive disorder characterized by arrest progressive mental and physical retardation, a prematurely aged appearance, microcephaly, ataxia, sensorineural deafness and bilateral congenital cataract [9,10]. Congenital Cataract-Facial Dysmorphism- Neuropathy (CCFDN) syndrome is an autosomal recessive syndrome that resembles Marinesco-Sjogren syndrome [11] . Smith-Lemli-Optiz syndrome is another autosomal recessive cause of infantile cataracts and microcephaly, which is suspected soon after birth, because of associated malformations including cleft palate, ptosis, ambiguous genitalia in males, polydactyly and malformations of the brain and kidneys [1 2]. Neu-Laxova syndrome is a further autosomal recessive cause of congenital cataracts and microcephaly but is easly recognized by the sloping forehead, micrognathia, protruding eyes often with absent lids, ichtyosis and absent hair, contractures, and syndactyly.
Fewer autosomal recessive cataract loci have been mapped. To date, 13 loci residing on chromosomes 1p34.4-p32.2, 1q21.1, 3p22-24.2, 6p23-24, 9q13-22, 16q21- 22, 19q13, 19q13.4, 20p12.1, 21q22.3, 22q11 , 22q12.1 and 17q, have been mapped, with six of these also causing autosomal dominant cataracts [1 3-26]. Of these loci, mutations in connexin 50 (GJA8), glucosaminyl (N-acetyl) transferase 2 (GCNT2), heat-shock transcription factor 4 (HSF4), lens intrinsic membrane protein (LIM2), beaded filament structural protein 1 (BFSP1), alpha A-crystallin (CRYαA), beta B1- crystallin (CRYβB1), beta B3-crystallin (CRYβB3), eph-receptor type-A2 (EPHA2) and galactokinase (GALK1) have been found [1 4,16,18,20-26]. None of these genes, although some of its are expressed both in the eye and brain, was reported responsible for an association between congenital cataract, GDD and microcephaly.
Conclusion
This is the first report of a new familial syndrome associating ARCC , GDD and microcephaly and it is of great interest to identify the genetic cause leading to such syndrome in children.
Declarations
Author contributions: Design and conduct of the study: CM.; collection of the data: C.M.,O.I., K.L; analysis and interpretation of the data, preparation, review, and approval of manuscript. C.M.
Conflict of interest: The authors of this article have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Institutional review board statement: Ethical approval was obtained from the Institutional Review Boards Committee of Charles Nicolle’s hospital.
Informed consent statement: Written consent was obtained from the patients.
Acknowledgments: The authors thank all the patients and their family members for participating in the project. This study was supported by the Tunisian ministry of higher education and scientific research (laboratory of human genetics Faculté de Médecine de Tunis, and congenital and hereditary service of Charles Nicolle’s hospital).
References
- Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008; 19: 134-49.
- Stoll C, Alembik Y, Dott B, Roth MP. Congenital eye malformations in 212, 479 consecutive births. Ann Genet. 1997; 40: 122- 128.
- Zetterström C, Lundvall A, Kugelberg M. Cataracts in children. J. Cataract Refract Surg. 2005; 31: 824-40.
- Jackson AP, Mc Hale DP, Compbell DA, Jafri H, Rashid Y, et al. Primary autosomal microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet. 1998; 63: 541- 546.
- Stromme P, Strokke O, Jellum E, Skjeldal OH, Baumgartner R. A typical methylmalonic aciduria with progressive encephalopathy, microcephaly and cataract in two siblings-a new recessive syndrome? Clin Genet. 1995; 48: 1- 5.
- Hennekam RC, Van de Meeberg AG, Van Doorne JM, Dijkstra PF, Bijlsma JB. Martsolf syndrome in a brother and sister: clinical features and pattern of inheritance. Eur J Pediatr. 1988; 147: 539-543.
- Warburg M, Sjo O, Fledelius HC, Perdersen SA. Autosomal recessive microcephaly, microcornea, congenital cataract, global developmental delay, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child. 1993; 147: 1309-1 312.
- Rainbow AJ, Howes M. A deficiency in the repair of UV and gamma-ray damaged DNA in fibroblasts from Cockayne’s syndrome. Mutat Res. 1982; 93: 235-247.
- Nance MA, Berry SA. Cockayne syndrome: Review of 140 cases. Am J Med Genet. 1992; 42: 68-84.
- Spivak G. The many faces of Cockayne syndrome. Proc Natl Acad Sci USA 2004; 101: 15273-1 5274.
- Varon R, Gooding R, Steglich C, Marns L, Tang H, et al. Partial deficiency of the C- terminaldomain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet. 2003; 35: 185-1 89.
- Nowaczyk MJ, Waye JS. The Smith-Lemli-Opitz syndrome: A novel metabolic way of understanding developmental biology, embryogenesis, and dysmorphology. Clin Genet. 2001; 59: 375-386.
- Butt T, Yao W, Kaul H, Xiaodong J, Gradstein L, et al. Localization of autosomal recessive congenital cataracts in consanguineous Pakistani families to a new locus on chromosome 1p. Mol Vis. 2007; 13: 1635-40.
- Ponnam SP, Ramesha K, Tejwani S, Ramamurthy B, Kannabiran C. Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet. 2007; 44: e85.
- Pras E, Pras E, Bakhan T, Levy-Nissenbaum E, Lahat H, et al. A gene causing autosomal recessive cataract maps to the short arm of chromosome 3. Isr Med Assoc J. 2001; 3: 559-562.
- Pras E, Raz J, Yahalom V, Frydman M, Garzozi HJ, et al. A nonsense mutation in the glucosaminyl (Nacetyl) transferase 2 gene (GCNT2): Association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci. 2004; 45: 1940-1 945.
- Heon E, Paterson AD, Fraser M, Billingsley G, Priston M, et al. A progressive autosomal recessive cataract locus maps to chromosome 9q13-q22. Am J Hum Genet. 2001; 68: 772-777.
- Smaoui N, Beltaief O, BenHamed S, M’Rad R, Maazoul F, et al. A homozygous splice mutation in the HSF4 gene is associated with an autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci. 2004; 45: 2716-2721.
- Riazuddin SA, Yasmeen A, Zhang Q, Yao W, Sabar MF, et al. A new locus for autosomal recessive nuclear cataract mapped to chromosome 19q13 in a Pakistani family. Invest Ophthalmol Vis Sci. 2005; 46: 623-626.
- Pras E, Levy-Nissenbaum E, Bakhan T, Lahat H, Assia E, et al. A missense mutation in the LIM2 gene is associated with autosomal recessive presenile cataract in an inbred Iraqi Jewish family. Am J Hum Genet. 2002; 70: 1363-1 367.
- Pras E, Frydman M, Levy-Nissenbaum E, Bakhan T, Raz J, et al. A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci. 2000; 41: 3511- 3515.
- Ramachandran RD, Perumalsamy V, Hejtmancik JF. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet. 2007; 121: 475-482.
- Riazuddin SA, Yasmeen A, Yao W, Sergeev YV, Zhang Q, et al. Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol Vis Sci 2005; 46: 2100-2106.
- Cohen D, Bar-Yosef U, Levy J, Gradstein L, Belfair N, et al. Homozygous CRYBB1 deletion mutation underlies autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci. 2007; 48: 2208-2213.
- Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis. 2010; 16: 511- 517.
- Yasmeen A, Riazuddin SA, Kaul H, Mohsin S, Khan M, et al. Autosomal recessive congenital cataract in consanguineous Pakistani families is associated with mutations in GALK1. Mol Vis. 2010; 16: 682-688.
