
Open Access, Volume 10
Beckwith-Wiedemann syndrome with Hirschsprung’s disease in a newborn case report and literature review
Ola Shahrour1; Alaa Mehair1; Ali Ahmed Alsaadi2; Aisha Al Shamsi3*
1Department of Academic Affairs, Tawam Hospital, Al Ain, United Arab Emirates.
2Laboratory Department, Clinical Scientist, Union 71, Tawam Hospital, Al Ain, United Arab Emirates.
3Pediatrics Department, Genetic Division, Tawam Hospital, Al Ain, United Arab Emirates.
Aisha Al Shamsi
Genetic Division, Pediatrics Department, Tawam Hospital, Al Ain, United Arab Emirates.
Email: aishamsi@seha.ae
Received : May 22, 2024,
Accepted : June 14, 2024
Published : June 20, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Background: Beckwith-Wiedemann Syndrome (BWS) is a genetic condition related to changes in the genes of chromosome 11 which result in the overgrowth of many parts of the body and are associated with multiple congenital anomalies. Hirschsprung’s disease is a congenital disorder characterized by a complete absence of ganglion cells in the myenteric and submucosal plexus of the colon. So far, only 2 patients with Beckwith-Wiedemann syndrome and intestinal obstruction due to Hirschsprung’s disease were reported.
Case presentation: We report an Emirati patient with a history of low birth weight, myoclonic seizure, subtle dysmorphic features, along with recurrent abdominal distension, and history of meconium plug syndrome for which further investigations were done including rectal biopsy which confirmed the diagnosis of Hirschsprung’s disease by the absence of ganglionic cells. His chromosomal microarray showed one copy gain of the size 963 kb on the short arm of chromosome 11 at the cytoband 11p15.5. The methylation study showed large duplication within the 11p15 imprinted region affecting imprinted domain H19/IGF2: IGDMR (IC1) confirming the diagnosis of Beckwith-Wiedemann syndrome.
Conclusion: This report adds to the genotype-phenotype correlation, highlighting the clinical importance of considering Hirschsprung’s disease as part of the manifestations of Beckwith-Wiedemann syndrome. As Early recognition of this association is crucial to provide effective treatment and decrease morbidity.
Keywords: Beckwith-Wiedemann syndrome; Hirschsprung’s disease; Microarray; Methylation study.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Al Shamsi A (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Shahrour O, Mehair A, Alsaadi AA, Al Shamsi A. Beckwith-Wiedemann syndrome with Hirschsprung’s disease in a newborn case report and literature review. Open J Clin Med Case Rep. 2024; 2257.
Introduction
Beckwith-Wiedemann Syndrome (BWS) is a growth disorder variably characterized by neonatal hypoglycemia, macrosomia, macroglossia, hemihyperplasia, omphalocele, embryonal tumors (e.g., Wilms tumor, hepatoblastoma, neuroblastoma, and rhabdomyosarcoma), visceromegaly involving one or more intra-abdominal organs including liver, spleen, kidneys, adrenal glands, and/or pancreas), renal abnormalities (e.g., medullary dysplasia, nephrocalcinosis, medullary sponge kidney, and nephromegaly), and anterior linear ear lobe creases and/or posterior helical ear pits. Sometimes there are characteristic facies including midface retrusion and infraorbital creases, vascular lesions including nevus simplex (typically appearing on the forehead, glabella, and/or back of the neck) or hemangiomas (cutaneous or extracutaneous), diastasis recti and advanced bone age. Other rare features include Cleft palate and cardiomyopathy [1]. Pregnancy-related findings were also reported including polyhydramnios, placental mesenchymal dysplasia and prematurity [2].
BWS is considered a clinical spectrum, in which affected individuals may have many of these features or may have only one or two clinical features. Early death may occur from complications of prematurity, hypoglycemia, cardiomyopathy, macroglossia, or tumors. However, the previously reported mortality of 20% is likely an overestimate given better recognition of the disorder along with enhanced treatment options. Macroglossia and macrosomia are generally present at birth but may have a postnatal onset. The growth rate slows around age seven to eight years. Hemihyperplasia may affect segmental regions of the body or selected organs and tissues [1].
The reported prevalence of BWS of 1:10,000 [3] to 1:13,700 [4] likely represents an underestimate given the existence of undiagnosed individuals with milder phenotypes. BWS has been reported in a wide variety of ethnic populations with an equal incidence in males and females.
A provisional diagnosis of BWS based on clinical assessment may be confirmed by molecular/ cytogenetic testing. Cytogenetically detectable abnormalities involving chromosome 11p15 are found in 1% or fewer of affected individuals. An epigenetic or genomic alteration leading to abnormal methylation at 11p15.5 or a heterozygous BWS-causing pathogenic variant in CDKN1C in the presence of one or more clinical findings [1]. Molecular genetic testing can identify epigenetic and genomic alterations of chromosome 11p15 in individuals with BWS:
- Loss of methylation on the maternal chromosome at Imprinting Center 2 (IC2) in 50% of affected individuals
- Paternal uniparental disomy for chromosome 11p15 in 20%
- Gain of methylation on the maternal chromosome at Imprinting Center 1 (IC1) in 5%.
Hirschsprung’s Disease (HD) has a classic triad of symptoms including abdominal distension, vomiting, and delayed passage of meconium. About one-third of patients can present with enterocolitis. It can occur as an isolated trait in 70% of cases.
Many case reports have documented the association of HD with various syndromes like down syndrome and others.
Reviewing the literature, we found 2 patients with Beckwith-Wiedemann syndrome and intestinal obstruction due to Hirschsprung’s disease [5,6], and here we are reporting a third case.
Materials and Methods
Ethical compliance: This study was approved by our regional research ethical committee. Ref. No.: MF2058-2022-900
Patient
An infant was evaluated by the Genetic team at Tawam Hospital, Al Ain, United Arab Emirates. Molecular characterization of the etiology was made after being evaluated. This patient was born to a nonconsanguineous parent (Figure 1). Blood samples from the family were collected in Ethylene Diamine Tetra Acetic Acid (EDTA) tubes after obtaining written informed consent.
Genetic evaluation
After written informed consent was obtained, blood samples were collected from the patient and the parent blood. Genomic DNA is purified, and quantified, from a patient sample (whole blood) using a QIAamp DNA Blood Mini kit (Qiagen). The DNA is then fragmented, amplified, and hybridized to the array according to the manufactural guidelines. CytoScan™ HD Array enables the detection of high-resolution Copy Number Variations (CNVs), large deletions, and/or duplications, across the genome. It also provides information about allelic imbalance from Single Nucleotide Polymorphisms (SNPs). It provides greater than 2.4 million markers for copy number and approximately 750,000 genotype-able SNPs across the genome. The Chromosome Analysis Suit (ChAS, Affymetrix) is used for the analysis of results. CNVs deletions that are greater than 50 kb, and duplications that are greater than 200kb were reported. Deletions of 25 consecutive markers or Duplication of 50 consecutive markers were reported. CytoScan™ HD Array SNP component allows the detection of Uniparental Disomy (UPD), loss (absence) of Heterozygosity AOH/LOH, parental consanguinity, and ploidy change. The results were interpreted using the following databases (solely or combinations): The Database of Genomic Variants (DGV), Decipher databases, OMIM and recent publications.
Clinical description
A male newborn was born at 31 weeks gestation to an Emirati couple, by normal spontaneous vaginal delivery in a private hospital with a birth weight of 1800 g. Antenatally was unremarkable except for His antenatal scan that showed dilated pelvis At birth, he had poor breathing with cyanosis and required resuscitation in the form of continuous positive airway pressure with high oxygen concentration (80%) to maintain oxygen saturation >90% and he was shifted to the neonatal Intensive unit for more care. Moreover, he had myoclonic seizures and was evaluated for it and was found to have sepsis due to E. coli for which he was treated with antibiotics.
At day 14 of life, he was transferred to our hospital for further evaluation and management. And Upon arrival at our hospital on day 14 of life, his weight was 2120 g (43th centile), length of 42 cm (17th centile), and head circumference of 29 cm (10th centile).
During the evaluation, he was noted to have some dysmorphic features in the form of down-slanting palpebral fissures, depressed nasal bridge, long smooth philtrum, and retrognathia (mother has the same). His head was small with overlapping sutures.
As his seizure was not well controlled, basic metabolic work up were done, all were unremarkable except plasma uric acid which was very low multiple times. He had high uric acid excretion. Urine for purine/pyrimidines profile was done and reported as normal. Due to this, the possibility of either renal or GI disease was suspected. His abdominal ultrasound showed bilateral hydroureteronephrosis.
Electroencephalogram showed epileptic changes; thus, he was started on antiepileptic medications including Levetiracetam and Phenobarbital. His brain MRI at 2 weeks of life showed relative flattening of the gyri of the frontal lobes bilaterally. This could be related to prematurity (early brain development). The cerebellum is relatively small in size compared to the rest of the supratentorial brain parenchyma (? Related to prematurity), no restricted diffusion. No signs of acute ischemic infarction or hemorrhage. No enhancing intracranial lesion is seen. No intracranial fluid collections. His brain MRS did not detect any abnormality.
During his stay in our hospital, he developed abdominal distention, feeding intolerance and constipation. Abdominal x-rays were done and showed distended dilated loops, no air in rectum, therefore, surgeons evaluated him to rule out any intestinal obstruction. Lower GI contrast was done and showed some areas of mucosal irregularity with no definite segmental colonic or rectal narrowing identified. As there was history of meconium plug, rectal biopsy was performed and showed absence of ganglion cells confirming Hirschsprung’s disease. His microarray was done for his dysmorphic features and seizure disorder, showed a likely pathogenic gain 11p15.5, region of Beckwith-Wiedemann syndrome and SilverRussell syndrome. Methylation study confirmed Beckwith-Wiedemann syndrome.
This patient was born to a non-consanguineous couple (Figure 1). Mother has some facial features similar to the index case and father has low IQ with schizophrenia and bipolar affective disorder. There was no history of miscarriages, neonatal deaths, infertility, developmental regression, or cancer in the family. Only significant history is that the father’s brother has son with macroglossia (not investigated).
Genetic analysis
Chromosomal Microarray (CMA) showed one copy gain of the size 963 kb on the short arm of chromosome 11 at the cytoband 11p15.5. The gain region involves 39 genes; 21 genes are reported in OMIM (BRSK2 (609236), MOB2 (611969), DUSP8 (602038), KRTAP5-1 (148022), IFITM10 (618293), CTSD (116840), SYT8 (607719), TNNI2 (191043), LSP1 (153432), TNNT3 (600692), MRPL23 (600789), H19 (103280), MIR675 (615509), IGF2 (147470), IGF2-AS (610146), INS (176730), TH (191290), ASCL2 (601886), C11orf21 (611033), TSPAN32 (603853), CD81 (186845)) (Figure 2).
The methylation study showed a large duplication within the 11p15 region affecting imprinted domain H19/IGF2: IG-DMR (IC1) detected by MS-MLPA. In addition, an aberrant methylation pattern (gain of methylation) at IC1 was detected. A normal methylation pattern was detected at the imprinted domain KCNQ1OT1: TSS-DMR (IC2). By this, genetic diagnosis of Beckwith-Wiedemann syndrome (BWS) due to a duplication on the paternally derived chromosome 11 is confirmed.
Parents had CMA, father has the same copy gain of the size 960 kb on the short arm of chromosome 11 at the cytoband 11p15.5 while the mother has a normal result.
Table 1: Summary of clinical phenotypes of the reported patients with BWS and Hirschsprung’s disease.
| Current report Patient 1 | Shah N, et al. (2020) Patient 2 | Cazzaniga L, et al. (2022) Patient 3 | |
|---|---|---|---|
| Age at exam/Gender | 14 days, Male | At birth, Female | 2.5 months, Male |
| Ethnic origin | United Arab Emirates | India | Italy |
| Consanguinity | None | None | None |
| Age at diagnosis | Neonatal | Neonatal | Post neonatal |
| Molecular confirmation | CMA showed gain at cytoband 11p15.5. Methylation showed large duplication within 11p15 region affecting imprinted domain H19/IGF2: IG-DMR (IC1), aberrant methylation pattern (gain of methylation) at IC1 was detected. |
Genetic analysis confirmed gain of methylation detected within detection limits of methylation- specific multiplex ligation-dependent probe amplification in H19 DMR (IC1) domain on 11p15 chromosome |
Genetic tests showed presence of mosaicism for loss of methylation of IC2. A correct pattern of methylation at IC1 locus was evident; presence of microdeletions or microduplications within 11p15.5 region excluded. |
| Polyhydramnios Placentomegaly Gestational age Large for gestational age Macroglossia Facial nevus simplex Ear creases/pits Abdominal wall defects: Omphalocele Umbilical hernia Diastasis recti |
No No 31 weeks No No No No No No No |
No No Full-term Yes Yes No Yes No Yes No |
Yes No 35 weeks Yes Yes No No No No No |
| Lateralized overgrowth/ hemihypertrophy |
No | Yes | No |
| Hypoglycaemia Transient Hyperinsulinism Pancreatectomy |
No | Yes No Yes Yes |
No |
| Organomeglay Hepatomegaly Nephromegaly Splenomegaly |
No Bilateral hydroureteronephrosis No |
No No No |
No Asymmetry of kidneys L>R No |
| Tumor | No | No | No |
| Others: Cleft palate Undescended testes Cardiac Hirschsprung’s disease Family history of BWS |
No No Normal Yes Yes, father |
No Not applicable Normal Yes No |
No No Patent foramen ovale (PFO) Yes No |
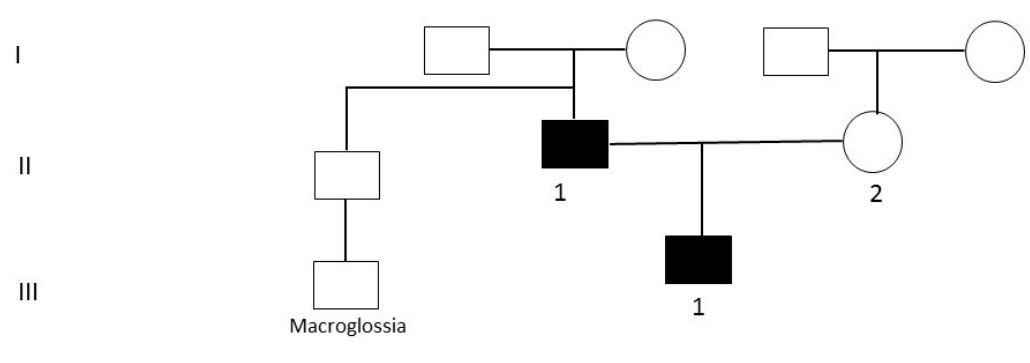
Figure 1: Family pedigree. Circles and squares are females and males, respectively; filled symbols are
affected members; Roman numbers indicate the generations; and Arabic numbers indicate offspring.
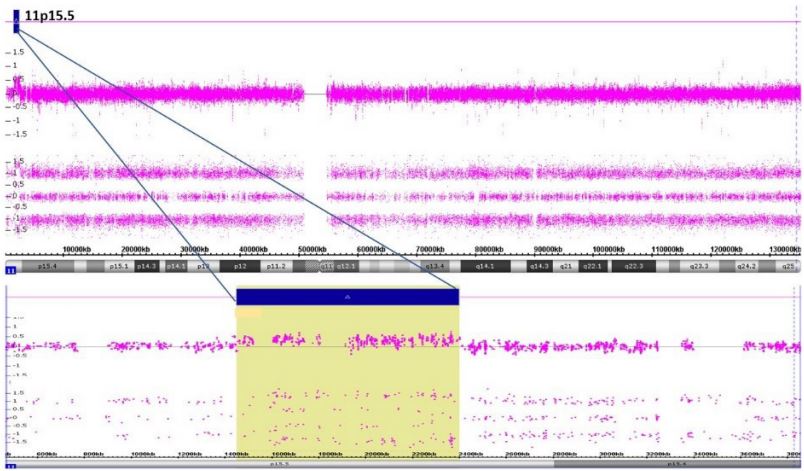
Figure 2: Chromosomal Microarray (CMA) showing one copy gain of the size 963 kb on the short arm
of chromosome 11 at the cytoband 11p15.5.
Discussion
Hirschsprung’s Disease (HD) has a classic triad of symptoms including abdominal distension, vomiting, and delayed passage of meconium. About one-third of patients can present with enterocolitis. Fecal stasis leads to the overgrowth of bacteria and mucosal inflammation, causing explosive diarrhea, abdominal distension, fever, and vomiting, along with leukocytosis. If left untreated, perforation of the bowel, septic shock, and death can result. Overall, 18% to 50% of patients with HD develop enterocolitis in their lifetime [7]. Hirschsprung’s disease occurs as an isolated trait in 70% of cases. Many case reports have documented the association of HD with various syndromes like Down syndrome, Bardet-Biedl syndrome, congenital central hypoventilation syndrome, familial dysautonomia, Goldberg-Shprintzen syndrome, MEN2A, and MEN2B syndromes and Smith-Lemli-Opitz syndrome [8,9].
On the other hand, BWS is an overgrowth syndrome, with patients often presenting with macroglossia, abdominal wall defects, hemihyperplasia, enlarged abdominal organs and an increased risk of embryonal tumors during early childhood. BWS is caused mainly by genetic or epigenetic defects within the chromosome 11p15.5 region. Thus, early recognition of the condition in the prenatal or neonatal period is critical for monitoring and timely treatment of complications [10].
To the best of our knowledge, the association of BWS with Hirschsprung’s disease has been reported only in two patients, and our patient is the third (Table 1 summarized the reported cases).
Our patient did not have the classical features of BWS, i.e. he did not have hypoglycemia or macroglossia but he presented with myoclonic seizure. Only two reports with female patients with BWS reported to have seizures [11,12]. The presence of persistently low plasma uric acid levels with a normal purine/pyrimidines profile was early indirect indication of either GI/renal pathology in our patient. Initially, bilateral hydroureteronephrosis was suspected to be the reason, but later on when he developed abdominal distension and feeding intolerance this was related to be the reason of the low plasma uric acid level.
It was known that the clinical manifestations of colon hypoganglionosis can be quite underhand and belatedly diagnosed; and the association between BWS and dysganglionosis could be rare [6], for that any early suspicion should take it into consideration and proceed with full genetic workup including Chromosomal Microarray (CMA) which can detect a deletion or duplication in a proband [1], keeping in mind that the ability to size the deletion depends on the type of microarray used and the density of probes in the 11p15.5 region, SNP array analysis can also detect segmental paternal uniparental disomy. CMA is considered first in a proband with intellectual disability as in our patient’s father.
Despite that approximately 85% of individuals with BWS have no family history of BWS; 15% have a family history consistent with parent-of-origin autosomal dominant transmission [1] as in our patient who inherited the syndrome from his affected father. Children of subfertile parents conceived by Assisted Reproductive Technology (ART) may be at increased risk for imprinting disorders, including BWS. Identification of the underlying genetic mechanism causing BWS permits better estimation of recurrence risk, and provide a prenatal screening for high risk pregnancies by specific prenatal testing either by CMA or methylation study for our patient’s family.
This report adds to the genotype-phenotype correlation, highlighting the clinical importance of considering Hirschsprung’s disease as part of the manifestations of Beckwith-Wiedemann syndrome. Early recognition of this association is crucial to provide effective treatment and decreases morbidity.
Declarations
Consent for publication: Consent for publication was obtained from the reported family.
Availability of data and materials: All data generated or analyzed during this study are included in this published article.
Competing interests: The authors declare that they have no competing interests
Funding: The authors disclosed that there is no financial support for the research, authorship, and/or publication of this article.
Authors’ contribution: Authors contributed to conception and design. All authors contributed to acquisition, revised manuscript and agreed to be accountable for all aspects of work ensuring integrity and accuracy. All authors read and approved the final manuscript.
Acknowledgement: The authors would like to thank the patients for taking part in this study and for giving permission to share their data.
References
- Shuman C, Beckwith JB, Weksberg R. Beckwith-Wiedemann Syndrome. [Updated 2016 Aug 11]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. 2000; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1394/
- Wilson M, Peters G, Bennetts B, McGillivray G, Wu ZH, et al. The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: Two new reports. Am J Med Genet. 2008; 146A: 137-148
- Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, et al. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am J Med Genet A. 2013; 161A: 2481-6.
- Thorburn MJ, Wright ES, Miller CG, Smith-Read EH. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child. 1970; 119: 316-21.
- Shah N, Khadilkar A, Khadilkar V, et al. Rare association of Beckwith-Wiedemann syndrome with Hirschsprung’s disease in an infant with hypoglycemia BMJ Case Reports CP. 2020; 13: e235121.
- Cazzaniga L, Parma B, Licini L, Dalla Rosa D, Cheli M, et al. Colon hypoganglionosis in Beckwith-Wiedemann syndrome: A new rare comorbidity? Clin Dysmorphol. 2022; 31(1): 18-22. doi: 10.1097/MCD.0000000000000390.
- Kroll-Wheeler L, Wilson AM. Educational Case: Hirschsprung Disease. Acad Pathol. 2019; 6: 2374289519893088. doi: 10.1177/2374289519893088.
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. Journal of Medical Genetics. 2008; 45: 1-14.
- Parisi MA. Hirschsprung Disease Overview. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Gene Reviews®. Seattle, WA: University of Washington. 2002; 1993-2019.
- Brioude F, Kalish J, Mussa, A. et al. Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018; 14: 229-249. https://doi.org/10.1038/nrendo.2017.166
- Kuzenkova LM, Kremenchugskaya MR, Globa OV, Podkletnova TV. Case of combination of Beckwith-Wiedemann syndrome with West syndrome. Vestn Ross Akad Med Nauk. Russian. 2014; 9-10: 64-9. doi: 10.15690/vramn.v69i9-10.1133.
- Mohan KR, Simalti AK, Kanitkar M. Cases of Beckwith-Wiedman Syndrome. Med J Armed Forces India. 2009; 65(2): 182-3. doi: 10.1016/S0377-1237(09)80142-1.
