
Open Access, Volume 10
Investigation of lipodystrophy in a 3-year-old and 6-month-old child: A case report
Natália Rossin Guidorizzi1*; Maria Cristina Foss-Freitas2*
1Brazilian Group for the Study of Inherited and Acquired Lipodystrophies (BRAZLIPO), Brazil.
2Department of Internal Medicine, Division of Metabolism, Endocrinology and Diabetes (MEND), Michigan Medicine, University of Michigan - Ann Arbor, MI, USA.
Natália Rossin Guidorizzi1*; Maria Cristina Foss-Freitas2*
1Brazilian Group for the Study of Inherited and Acquired Lipodystrophies (BRAZLIPO), Brazil.
Tel: +55-19-99191-2470;
Email: nrguidorizzi@hcrp.usp.br
2Department of Internal Medicine, Division of Metabolism, Endocrinology and Diabetes (MEND), Michigan
Medicine, University of Michigan - Ann Arbor, MI, USA.
Tel: +1-734-615-6253;
Email: mfossdef@med.umich.edu
Received : Mar 12, 2024,
Accepted : Apr 25, 2024
Published : Apr 30, 2024,
Archived : www.jclinmedcasereports.com
Abstract
This report delineates the case of a 3-year-old and 6-month-old child exhibiting noteworthy weight loss post-vaccination, succeeded by modest adipose tissue gain and subsequent phenotype deterioration following a viral episode. Additionally, the patient presents phlebomegaly, muscular hypertrophy, and irritability linked to hyperphagia. Despite extensive investigation encompassing celiac disease exclusion, the prevailing diagnostic hypothesis is acquired generalized lipodystrophy.
Keywords: Lipodystrophy; Weight loss; Hyperphagia; Muscular hypertrophy; Phenotype.
Abbreviations: APLV: Allergy Protein Milk Cow’s; DM1: Type 1 Diabetes Mellitus; HAS: Hypertension Arterial Systemic.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Rossin Guidorizzi N & Foss-Freitas MC (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Guidorizzi NR, Foss-Freitas MC. Investigation of lipodystrophy in a 3-year-old and 6-month-old child: A case report. Open J Clin Med Case Rep. 2024; 2233.
Introduction
Lipodystrophy, a rare condition characterized by selective, localized, partial, or nearly generalized loss of subcutaneous adipose tissue, presents both acquired and congenital forms. Acquired lipodystrophy may manifest in tandem with various conditions, including viral infections, autoimmune diseases, metabolic disorders, and adverse drug reactions. This case report elucidates a child with acquired generalized lipodystrophy whose clinical manifestation and progression posed diagnostic challenges.
Case Presentation
The patient, a 3-year-old and 6-month-old girl, delivered via cesarean section with a birth weight of 3600 g and length of 51 cm, displayed a typical adipose tissue distribution. Despite experiencing cow’s milk protein allergy necessitating infant formula due to inadequate breastfeeding, she exhibited appropriate weight, height gain, and neuropsychomotor development for her age. Following poliomyelitis vaccination at one year and three months, she underwent rapid and significant weight loss (approximately 1.7% in a few days), primarily in the limbs, followed by modest weight gain while maintaining reduced adipose tissue in the areas above. However, after a viral episode in October 2023, a deterioration in phenotype ensued, characterized by phlebomegaly, muscular hypertrophy, and irritability associated with hyperphagia (Figure 1). Extensive etiological investigation revealed hypertriglyceridemia, hepatomegaly with hepatic steatosis, altered transaminases, and hyperinsulinemia. Leptin levels were undetectable, Complement 4 (C4) levels below the lower limit of normality, and viral infections such as HIV and hepatitis B and C were ruled out. The patient did not present hematological neoplasms, undergo bone marrow transplantation, or use checkpoint inhibitors.
Furthermore, she exhibited no autoimmune diseases. Genetic testing for candidate lipodystrophy genes yielded no suggestive genes. The parents, although non-consanguineous and devoid of a similar phenotype, had the father diagnosed with type 1 diabetes at age 4.
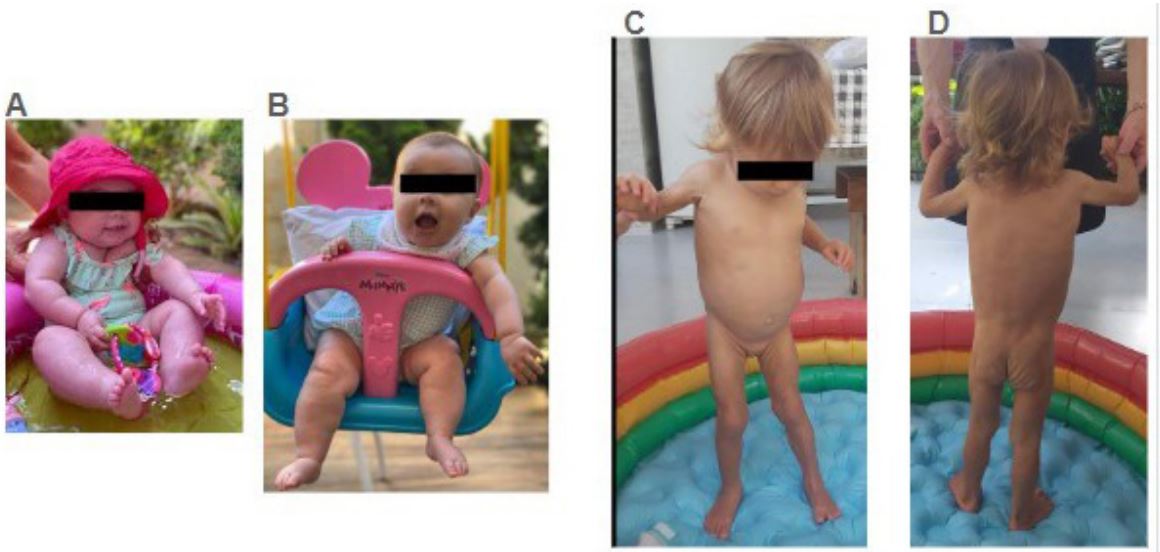
Figure 1 and 2: Pre-operative implants XR and CT scan.
Discussion/Conclusion
Acquired generalized lipodystrophy, or Lawrence Syndrome, represents a rare lipodystrophic syndrome characterized by adipose tissue loss, insulin resistance, and an elevated cardiovascular risk. Over 100 cases have been documented, with a female-to-male ratio of 3:1. The clinical presentation mirrors that of Berardinelli-Seip syndrome, suggesting an acquired syndrome. Differential diagnoses include other forms of extreme insulin resistance and various lipodystrophies. Management primarily addresses metabolic manifestations akin to other insulin resistance syndromes, with treatment modalities such as lifestyle modifications, insulin sensitizers, antihypertensives, and hypertriglyceridemia management. Recombinant human leptin has demonstrated efficacy in some cases. The prognosis remains uncertain, likely influenced by cardiovascular risk and underlying etiology. Lawrence syndrome, or Acquired Generalized Lipodystrophy (AGL), signifies a rare acquired condition characterized by substantial fat loss across multiple body regions, notably the face, arms, and legs. While sharing similarities with other lipodystrophies, the etiology and progression of AGL remain distinct. This case underscores the diagnostic complexity of lipodystrophy in children, emphasizing the necessity for a multidisciplinary approach to ensure optimal growth and development [1-7].
Declarations
Acknowledgements: The authors thank the patient’s family for their collaboration and consent to share this case.
Financial support: There was no financial support.
Competing interests: Authors state no conflict of interest relevant to this manuscript.
Availability of data and material: Original data generated and analyzed are available through request to the corresponding authors.
Disclosure statement: All authors have nothing to disclose with respect to this manuscript.
References
- Foss-Freitas MC, Akinci B, Luo Y, Stratton A, Oral EA. Diagnostic strategies and clinical management of lipodystrophy. Expert Rev Endocrinol Metab. 2020; 15(2): 95-114. doi:10.1080/17446651.2020.1735360.
- Brown RJ, Araujo-Vilar D, Cheung PT, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016; 101(12): 4500-4511. doi: 10.1210/jc.2016-2466.
- Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J Endocrinol Invest. 2019; 42(1): 61-73. doi:10.1007/s40618-018-0887-z.
- Gonzaga-Jauregui C, Ge W, Staples J, et al. Clinical and molecular prevalence of lipodystrophy in an unascertained large clinical care cohort. Diabetes. 2020; 69(2): 249-258. doi:10.2337/db19-0447.
- Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes, Metabolic Syndrome and Obesity. 2017; 10: 375-383. doi:10.2147/dmso.s130810.
- Lightbourne M, Brown RJ. Genetics of Lipodystrophy. Endocrinol Metab Clin North Am. 2017; 46(2): 539-554. doi:10.1016/j.ecl.2017.01.012.
- Garg A. Lipodystrophies: Genetic and acquired body fat disorders. Journal of Clinical Endocrinology and Metabolism. 2011; 96(11): 3313-3325. doi:10.1210/jc.2011-1159.
