
Open Access, Volume 10
Non-dravet syndrome epilepsy in 4 polish patients with SCN1A de novo mutations
Anita Zielińska1; Paulina Górka-Skoczylas2; Dorota Hoffman-Zacharska2; Renata Tataj2; Anna Winczewka-Wiktor3; Tomasz Mazurczak1; Elżbieta Stawicka1*
1Institute of Mother and Child, Clinic of Paediatric Neurology, Poland.
2Department of Medical Genetics, Institute of Mother and Child, Poland.
3Department of Developmental Neurology, Poznan University of Medical Sciences, Poland.
Elżbieta Stawicka
Institute of Mother and Child, Clinic of Paediatric Neurology, Poland.
Email: elzbieta.stawicka@imid.med.pl
Received : Feb 01, 2024,
Accepted : Mar 07, 2024
Published : Mar 11, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Background: The spectrum of neurological disorders associated with mutations in SCN1A, which encodes the type 1 subunit of the neuronal voltage-gated sodium channel, is broad and can manifest as both epileptic (a heterogeneous group of epileptic syndromes) and non-epileptic conditions. Dravet syndrome (DRVT) and Genetic Epilepsy with Febrile Seizures plus (GEFS+) are the most frequent. However, there exist less common phenotypes with varying severity of clinical course that authors would like to highlight.
Case presentation: We would like to present the clinical descriptions of our four patients confirmed to have mutations in the SCN1A gene, presenting the least frequently described phenotypes. The first was initially diagnosed with ASD (Autism Spectrum Disorder) and Febrile Seizure plus (FS+), the second met the criteria for Lennox-Gastaut Syndrome (LGS), the third was diagnosed with Malignant Migratory Partial Seizures of Infancy (MMPSI), and the fourth with SCN1A-related Early Infantile Developmental and Epileptic Encephalopathy (EIDEE) without movement disorders. We described the course of the disease to date, the current status of the patients, and their main problems and referred to the type of pathogenic variants in the SCN1A gene.
Conclusion: The spectrum of phenotypes associated with mutations in the SCN1A gene is vast and our cases present this heterogeneity. We described a patient with febrile FS+ and an average course of the disease, two patients with severe and one with a highly severe course of illness. It is crucial to recognize that within the GEFS+ syndrome, there may exist phenotypes that are not necessarily familial or hereditary.
Not only in patients with DRVT or GEFS+ spectrum but also in those with other epileptic syndromes such as LGS, MMPSI, or DEE, the possibility of SCN1A-dependent etiology should be taken into account.
Keywords: Dravet syndrome; SCN1A gene; Non-dravet phenotype.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Stawicka E (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Zielińska A, Górka-Skoczylas P, Hoffman-Zacharska D, Tataj R, Winczewka-Wiktor A, et al. Non-dravet syndrome epilepsy in 4 polish patients with SCN1A de novo mutations. Open J Clin Med Case Rep. 2024; 2208.
Introduction
Mutations in the SCN1A gene are associated with a broad spectrum of children’s neurological disorders, encompassing both paroxysmal (a heterogeneous group of epileptic syndromes) and non-epileptic phenotypes. Among epileptic syndromes, the Dravet syndrome (DRVT, OMIM 607208) is the most common, but this group include disorders ranging in severity from relatively mild familial febrile seizures and Genetic Epilepsy with Febrile Seizure Plus (FEB3A, GEFSP2/GEFS+; OMIM 604403), to entities that are much more prognostically serious and categorized as developmental and epileptic encephalopathy 6B, non-Dravet type (DEE6B, OMIM 619317). Among non-epileptic disorders, Familial Hemiplegic Migraine type 3 (FHM3, OMIM 609634) has only been described. Seldom, SCN1A pathogenic variants are also identified in other forms of DEEs, including Myoclonic-Astatic Epilepsy (MAE, Doose syndrome), infantile epilepsy with migratory focal seizures (MMPSI), West Syndrome (WS) and Lennox-Gastaut Syndromes (LGS). [1,2].
The SCN1A gene (OMIM 182389), located in locus 2q24.3, encodes the alpha subunit of a voltagegated sodium channel (Nav1.1) that is crucial for generation and propagation on action potential - neural transmission in the central nervous system. The presentation of SCN1A-related disorders correlates with the type of pathogenic variants and the underlying change in functional properties (loss- or gain-of-function, LOF/GOF) of channels. LOF variants in the SCN1A gene cause DRVT and GEFSP2; while GOF variants are associated with FHM3 and the group of non-Dravet DEEs [3].
In this article, we would like to present clinical reports of four patients with mutations in the SCN1A, classified as pathogenic but presenting the least frequently described phenotypes: ASD and FS+ syndrome, the Lennox-Gastaut syndrome, MMPSI and EIDEE. The article highlights the importance of detailed characteristics of genotypic-phenotypic variability and its clinical relevance in daily clinical practice, even in the case of well-known genes such as SCN1A.
Methods
Clinical data was sourced from available medical records and interviews with parents. Molecular analysis of the SCN1A gene was performed by direct Sanger sequencing of coding exons or target NGS. SCN1A deletion/duplications were also excluded in the case of missense mutations.
Patient 1 (P1) 12-year-old boy with no family history of neurological disorders. He was born from a second pregnancy, natural birth, with an Apgar score of 10, his neonatal period was uncomplicated. In the fifth month of age, he had the first tonic-clonic seizure during fever - simple febrile seizures were diagnosed. Subsequent seizures were exclusively tonic-clonic with fever. Extensive diagnostics were performed: brain MRI imaging and metabolic diagnostics were normal. EEG recordings varied over time but were often within the age norm, with occasional small, generalized sharp waves during sleep. In the first year of life, antiepileptic treatment was started with Valproic Acid (VPA), followed by Topiramate (TPM). Seizures with fever continued. The boy’s psychomotor development was slightly delayed in gross motor skills with normal development of the cognitive sphere and speech. At the age of 3 years, SCN1A analysis was performed (Sanger sequencing) and a heterozygous nonsense variant - p.Trp952Ter (c.2855G>A), of de novo origin, was detected. Currently, the boy has an average of 1-2 seizures per year, constantly occurring with fever. He was treated with valproic acid, with significant improvement in psychomotor development after discontinuation of topiramate. No gross motor abnormalities, no ataxia, typical gait pattern for Dravet syndrome (crouching gait) was observed. The boy has been diagnosed with atypical autism, mild mental retardation, he attends a school therapy center. He receives additional physical rehabilitation. Currently the biggest problem is hyperactivity and attention deficits.
Patient 2 (P2) 15-year-old boy with no family history of neurological disease, born from second pregnancy delivered via caesarean section due to lack of labor progress. His Apgar score was 9-10. The neonatal period was uncomplicated. Severe esophageal reflux was observed in infancy, Sanger syndrome was suspected. The patient had normal psychomotor development until four months of age – he was turning sideways, catching toys, and smiling. First epileptic seizure occurred at four months of age, it was a focal clonic seizures, varying from side to side with postictal paresis. Since the onset of seizures, there has been a severe developmental delay. The boy started to sit up unaided at 24 months of age and to walk independently at the age of 2.5 years. Extensive neurological diagnostics were performed - a triple brain MR imaging- normal, metabolic tests- negative. At two years of age, SCN1A analysis (Sanger sequencing) revealed a heterozygous, frameshift variant p.Asp592MetfsTer31 (c.1773del), of de novo origin. During this period, atypical absence seizures, multiple epileptic states of tonic-clonic seizures, atonic seizures, myoclonic seizures occurred daily, focal tonic seizures every night. The EEG was described with abnormal basal function, multifocal lesions, and slow spike-wave activity of 1.5-3 Hz, usually in the frontal region. During infection, a reduction in seizure frequency was observed in the boy from infancy. Currently, the boy does not speak, has many motor stereotypies, and is extremely hyperactive. He attends an educational center and enjoys contact with other people. He is currently being treated with VPA, Stiripentol (STR), and quetiapine. The biggest current problem, apart from epileptic seizures, is sleep disturbance. The patient can stay awake for 50 hours (2-4 times a month before using quetiapine even three times more often), but sleep is constantly interrupted, lasting continuously for 3 to 4 hours.
Patient 3 (P3) A 7-year-old girl with a family history burdened by epilepsy (cousin treated with VPA), autism (the same cousin), and migraine (paternal grandmother). Born from second pregnancy by caesarean section at 38 weeks gestation. The pregnancy history was complicated by gestational diabetes - treated with insulin and high risk of premature labor treated with lutein. She scored 10 on the Apgar scale, but increased muscle tone was noted during neonatal examination. Her first focal seizures occurred at 3.5 months of age - unilateral clonic, followed by focal with disturbance of consciousness, unclassified, bilateral tonic-clonic and status epilepticus. Seizures were persistent, and no atonic or absence seizures were observed. EEG recordings up to 12 months were normal, but subsequent EEGs revealed various abnormalities, including spike/sharp wave, spike/slow wave, and spike/slow wave discharges, primarily over the left hemisphere during sleep, with periodic single responses on the right side. Discharges had disappeared during deep sleep. At 13 months: discharges localized mainly in the right cerebral hemisphere and generalized paroxysmal, almost continuous during sleep, at 18 months: multifocal lateralized (with hemispheric variability) and generalized paroxysmal discharges, continuous during sleep, at 34 months - bioelectrical status epilepticus. In the course of diagnosis, genetic testing was performed - tNGS using the panel of 49 EIEEs/DEEs genes (EIEE v.1/2016: http://zgm.imid.med.pl/panele-ngs/) and a heterozygous missense variant p.Ala1669Thr (c.5005G>A) of de novo origin was identified. Normal development was observed until the age of 14 months, then the development delayed (sitting at 10 months, crawling at 11 months, standing at 12 months, walking at 13 months near furniture, grasping with tweezers with independent placing of pieces of food in the mouth at 9 months, single two-syllable words at 12 months). At the age of 18 months generalized hypotonia was observed with continuous loss of motor functions. Currently, the girl does not sit, walk, or speak, is fed by PEG, and sleeps well at night. Parents observe breaks in seizures during infections. Since July 2023, when fenfluramine was introduced, the intervals between seizures have lengthened up to 19-23 days.
Patient 4 (P4) A 10-year-old boy with no family history of neurological diseases. Born from second pregnancy, natural delivery, scored 10 points on the Apgar scale. In the neonatal period, the first tonicclonic epileptic seizure occurred during an infection with high fever. Later, polymorphic seizures (mostly tonic-clonic) were observed not only due to infection. The boy was treated with various combinations of antiepileptic drugs (Table 1). Extensive diagnostics were performed, including brain MR imaging, which was normal, and metabolic tests, which was negative. Seizures continued to occur once a month on average. The boy’s psychomotor development in gross motor skills was extremely delayed- the boy has never sat or crawled. In the area of speech development - he does not speak or nonverbally communicate. At the age of 2.5 years, a genetic test for a mutation in gene SCN1A – direct sequencing of coding exons was performed, and heterozygous missense variant p.Ala239Asp (c.716C>A), of de novo origin, was identified. Because of the disease course, the second broader test was performed at the age of 7 years - tNGS using the panel of 49 EIEEs/DEEs genes (EIEE v.1/2016: http://zgm.imid.med.pl/panele-ngs/). SCN1A missense was confirmed, and no other pathogenic/likely pathogenic was identified. However, in the meantime, this variant has been annotated as pathogenic in HGMD Professional (CM208235) as causative for patients classified in the group early infantile-onset DEE (1 month < seizure onset ≤3 months), with seizure onset on 90 days [4]. Currently, the boy has tonic-clonic seizures once a week. The boy has a profound intellectual disability and attends a school-therapy centre. He also receives physical rehabilitation. Currently, according to his parents, his most significant problems are difficulties with feeding - he eats only mixed food and has problems with swallowing and communication. In the opinion of the teachers, there is a lack of cooperation.
Simplified pedigrees (FamP1-P4) indicate mutations de novo origin in all cases, Sanger sequencing chromatograms are presented for all variants. Created with BioRender.com. Source: Authors’ elaboration.
Table 1: Clinical comparison of patients
| P1 | P2 | P3 | P4 | |
|---|---|---|---|---|
|
Positive history of neurological diseases |
No | No |
Cousin treated in childhood
for epilepsy, migraine in paternal grandmother |
No |
|
Complications of the pregnancy period |
No complications |
Heart rate declines- caesarean section |
Pregnancy: diabetes treated
with insulin, preterm contractions |
No complications |
|
First seizure (months old) |
5 | 4 | 3,5 | 1 |
|
Psychomotor development |
Delayed in the sphere of gross motor skills |
Psychomotor development significantly delayed; at present, atactic gait and numerous motor stereotypies, does not speak |
Psychomotor development significantly delayed- does not sit, does not walk, does not speak, grouses, fed with PEG |
Psychomotor development significantly delayed- does not sit, does not walk, does not speak, grouse, and is fed orally with fragmented food |
| Type of seizures | Tonic-clonic |
Atypical absence seizures, multiple epileptic states of tonic- clonic seizures, atonic seizures, myoclonus, focal seizures |
Focal variable as to
side, focal with disturbance of consciousness, unclassified, bilateral tonic-clonic, status epilepticus, and transient myoclonus during treatment with levetiracetam |
Tonic-clonic |
| Treatment used |
Valproinian acid, topiramate |
Carbamazepine, valproic acid, vigabatrin, topiramate, clobazam, ethosuximide, levetiracetam, stiripentol, perampanel ,zonisamide, potassium bromide, steroid therapy |
Clonazepam, valproic
acid, ethosuximide, immunoglobulins, perampanel, carbamazepine, nitrazepam, stiripentol, trazodone, fenfluramine |
Phenobarbital,
topiramate, valproic acid, clobazam, stiripentol |
|
Neurological condition |
Mild intelectual disability, atypical autism |
Severe intelectual disability |
Profound intelectual disability |
Profound intelectual disability |
Table 2: Description and characterization of mutation identified in presented patients.
| ID |
c.DNA (NM_001 165963) |
Protein (NP_0011 59435.1) |
Inheritence |
Variant consequence |
Localization Exon/ Protein domain |
Functional prediction# |
Phenotype prediction## |
ACMG classification |
Paralogous pathogenic variant |
gnomAD variant freq. |
ClinVar/ HGMD* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | c.2855G>A | p.Trp952Ter | de novo | Stop gained | Ex18/ DIIS5-S6 |
LOF | DRVT (98.21%) | Pathogenic | - | 0 | DEE/ SMEI |
| P2 | c.1773del |
p.Asp592 MetfsTer31 |
de novo | Frameshift | Ex14/L1 | LOF | DRVT (98,89%) |
Likely Pathogenic |
- | 0 | - / - |
| P3 | c.5005G>A | p.Ala1669Thr | de novo | Missense | Ex29/ DIVS4-S5 |
LP (0.94)/ GOF (0.82 |
DRVT (95,15%) |
Likely Pathogenic |
SCN8A/ DEE, UE p.Ala1650Thr/ Val |
0 |
DEE/ DD, poor coordination and seizures |
| P4 | c.716C>A | p.Ala239Asp | de novo | Missense | Ex09/ DIS4-S5 |
LP (0.93)/ GOF (0.62) |
DRVT (98,78%) |
Likely Pathogenic |
SCN2A/DEE p.Ala240Ser |
0 | GEFS+/DRVT |
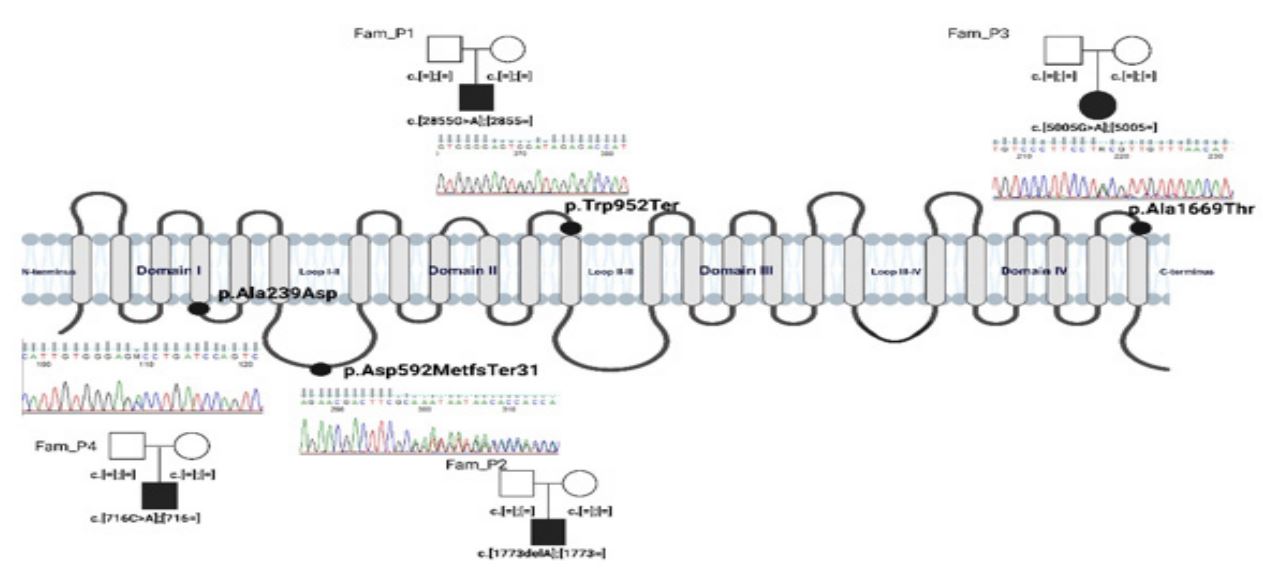
Figure 1: Schematic localization of SCN1A/Nav1.1 variants under analysis.
Discussion
We present four patients with pathogenic de novo mutations in the SCN1A gene who have not been diagnosed with Dravet Syndrome due to partial compliance of the clinical picture with the diagnostic criteria for DRVT.
The mandatory criteria for DRVT according to ILAE are: [5]
1) Recurrent focal clonic (hemiclonic) febrile and afebrile seizures (which often alternate sides from seizure to seizure), focal to bilateral tonic-clonic, and/or generalized clonic seizures,
2) Age of onset 1-20 months of life,
3) Drug-resistant epilepsy,
4) Intellectual disability
Also, according to C. Wirel et al. [6], DRVT can be diagnosed in a child aged 2-15 months who have had a single prolonged hemiclonic seizure or focal or generalized status epilepticus (≥30 min) of unknown etiology in the context of vaccination or fever.
In our group, the first seizures were typical for DRVT, as tonic-clonic seizures (P1,P4) and focal clonic seizures variable from side to side, prolonged in P2 and P3. In 3 of 4 patients, epilepsy is drug-resistant. All patients have intellectual disability, 3 of them preceded by normal psychomotor development in early infancy
Autistic features that are inherent in the clinical picture of patients with Dravet Syndrome were present in our group in varying degrees. P1 was diagnosed with atypical autism, while P2 with ASD with particularly severe stereotypic motor symptoms. In P3 and P4, the diagnosis in this area was not carried out, due to the fact that intellectual disability of profound degree was already known. Autistic signs have recently been reported in patients with DVRT, ranging from 8-61%, with verbal communication difficulties and impaired adaptive skills particularly highlighted [7].
Our patients show motor impairments ranging from mild delay in motor skills to complete inability to sit, with only the ability to stabilise the head (P3,P4). Kalume et al. [8] explain the motor impairments associated with mutations in the SCN1A gene by impaired expression of sodium channels not only in Purkinje cells, which is responsible for ataxia, but also in the nodes of Ranvier, including about 80% of motor neurons. This clarifies the distal motor deficit with gait disturbance, as well as the proximal one, leading to crouch.
The P1 did not meet the criteria for drug-resistant epilepsy and had only seizures provoked by hyperthermia. He responded very well to valproic acid, which does not comply with the criteria of DRVT. He only required the addition of a second drug during a prolonged tonic-clonic seizure throughout infection with fever. However, after the drug was discontinued, seizures did not aggravate. According to ILAE criteria [5] this patient’s phenotype aligns with the spectrum of GEFS+. As it is known, the most common symptom of the above spectrum is febrile seizures, but febrile seizures plus, which are present in the case described, are also frequently observed [9]. A first episode under six months of age and the necessity for chronic anticonvulsant treatment are not exclusion criteria [5,10]. Also, the literature has already described the lack of family burden and the type of mutation (de novo variant) [11,12].
In the P2, due to typical changes in the EEG signal (sleep-provoked bursts of slow spike and wave discharges and paroxysms of fast activity with the highest amplitude over the frontal region), the presence of tonic seizures during sleep and intellectual disability the LGS was diagnosed. Several authors [13,14] emphasize molecular diagnostics’ role in LGS patients, especially those with the myoclonic subtype. This is especially important to avoid using medications that can aggravate the patient’s state of health.
An additional problem for the patient includes sleep disorders. Insomnia significantly increases the frequency of epileptic seizures in majority of patients and negatively affects the quality of life of the child and family. The number of studies on this subject is limited. Papale et al. [15] performed an immunochemical tests of SCN1A gene expression in critical brain areas responsible for proper sleep architecture confirmed extensive SCN1A expression in the neocortex, hippocampus, hypothalamus, thalamic reticular nuclei, dorsal sutural nuclei, pedunculopontine nuclei and laterodorsal tegmental nuclei, which may have a direct impact on sleep disorders in patients with mutations in the SCN1A gene. The problem requires further studies. In a clinical trial using melatonin in patients with DRVT, there was no significant effect on sleep improvement. However, most caregivers reported a crucial effect on sleep onset and maintenance, corresponding enhancement of cognitive function and quality of life [16].
The P3 met the clinical criteria for DRVT. However, due to the characteristic abnormalities on EEG examination and a very severe phenotype, a diagnosis of MMPSI seemed more adequate. The MMPSI phenotype was diagnosed according to the following criteria: onset under 6 months; presentation with focal or hemiclonic seizures, with progression to frequent, almost continuous, multifocal seizures; developmental arrest at seizure onset and progressive cognitive decline with clusters of seizures [17]. That patient had the worst response to treatment. There was a reduction in numerous focal seizures (several per day) after initially using trazodone (because of sleep problems), after which the seizures returned. The patient is now treated with fenfluramine added to her treatment. The seizure reduction has occurred from the first dose, and she has achieved a substantial reduction in seizures and a significant increase in the time between seizures – at the beginning of treatment 19 to 23 days of seizure-free. Now, seizures occur on average every 10 days. This may indicate the effectiveness of this drug not only in DRVT patients. During treatment, carbamazepine was introduced for a short time, discontinued because of a significant regression of speech. Due to the type of mutation (GOF variant), a new trial of treatment with a sodium channel blocker is planned.
Rojo et al. [17] searched for a genetic cause in 15 patients with MMPSI by performing a test for mutations in genes associated with epileptic encephalopathies. In one patient, a mutation in the SCN1A gene was found. According to the authors [17], MMPSI encephalopathy associated with a mutation in the SCN1A gene is the most severe phenotype known. MMPSI was previously a diagnosis of unknown etiology, but now, with the dynamic development of molecular diagnostics, pathogenic variants have been described in about 70% of patients [18-22]. It is a syndrome with a significant genetic heterogeneity. Based on Zhang et al. [23], SCN1A is now being considered to be the third most frequent type of genetic variation in the MMPSI.
In the last Patient (P4), the first seizures occurred before he was one month old, and the psychomotor development was delayed from birth. Due to the drug-resistant course of illness, severe intellectual disability, serious motor disability, and the result of a genetic testing (GOF variant), the patient’s phenotype probably corresponds to that described by Brunklaus [24] as Early Infantile Developmental and Epileptic Encephalopathy (EIDEE) associated with the mutation of SCN1A gene. This study, which has been confirmed by other scientists [25-29] highlights the possibility of using medications that have been so far contraindicated in patients with DRVT, which are sodium blockers. Due to its limitations, the study guides the direction of further work while providing opportunities for introducing more effective, precise pharmacotherapy
Conclusion
The spectrum of phenotypes associated with mutations in the SCN1A is constantly expanding and is characterized by significant heterogeneity of the clinical course [28,29]. The descriptions of patients above illustrate this diversity, representing atypical, infrequent phenotypes. Although it is known that SCN1A is the leading cause of DVRT and Genetic Epilepsy with Febrile Seizures plus (GEFS+), clinicians should be aware of less common phenotypes.
According to the GEFS+ syndrome, it is essential to consider that there may be phenotypes that are not necessarily familial or hereditary.
Close teamwork between geneticists and clinicians is of utmost importance. The clinical diagnosis should not be determined solely based on gene sequencing but needs to be individualized according to the patient’s clinical symptoms to develop the optimal treatment regimen. However, the functional evaluation resulting from a specific mutation (the GOF or LOF variant) might be clinically relevant since it critically impacts the therapeutic decisions and the prognosis. Further studies are needed to understand the genotype-phenotype correlation in patients with SCN1A-dependent disorders. In the years ahead, therapy dedicated to a specific diagnosis, such as DRVT, might be replaced by personalized therapy accounting for an individual’s molecular profile.
References
- Scheffer IE, Nabbout R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia. 2019 ; 60 Suppl 3: 17-24.
- Ding J, Li X, Tian H, Wang L, Guo B, Wang Y, et al. SCN1A Mutation-Beyond Dravet Syndrome: A Systematic Review and Narrative Synthesis. Front Neurol. 2021; 12: 743726.
- Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology. 2017; 89(10): 1035-1042.
- Na JH, Shin S, Yang D, Kim B, Kim HD, Kim S, et al. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. 2020; 42(6) :438-448.
- Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63(6): 1349-1397.
- Wirrell EC, Hood V, Knupp KG, Meskis MA, Nabbout R, Scheffer IE, et al. International consensus on diagnosis and management of Dravet syndrome. Epilepsia. 2022; 63(7): 1761-1777.
- Ouss L, Leunen D, Laschet J, Chemaly N, Barcia G, Losito EM, et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: Delineation of a specific phenotype. Epilepsia Open. 2019; 4: 40–53.
- Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007 ;27(41): 11065-74.
- Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalised epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol. 1999; 45: 75–81.
- Scheffer IE, Berkovic SF. Generalised epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997; 120(Pt 3): 479–90.
- Bonanni P, Malcarne M, Moro F, Veggiotti P, Buti D, Ferrari AR, et al. Generalised epilepsy with febrile seizures plus (GEFS+): clinical spectrum in seven Italian families unrelated to SCN1A, SCN1B, and GABRG2 gene mutations. Epilepsia. 2004; 45: 149–58.
- Myers KA, Burgess R, Afawi Z, Damiano JA, Berkovic SF, Hildebrand MS, et al. De novo SCN1A pathogenic variants in the GEFS+ spectrum: not always a familial syndrome. Epilepsia. 2017; 58: e26-30.
- Selmer KK, Lund C, Brandal K, Undlien DE, Brodtkorb E. SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy Behav. 2009 ;16(3): 555-7.
- Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007; 843-852.
- Papale LA, Makinson CD, Ehlen C, Tufik S, Decker MJ, Paul KN, et al. Altered sleep regulation in a mouse model of SCN1Aderived genetic epilepsy with febrile seizures plus (GEFS+). J Clin Sleep Med. 2018 ;14(10): 1697-1704.
- Myers KA, Davey MJ, Ching M, Ellis C, Grinton BE, Roten A, et al. Randomised Controlled Trial of Melatonin for Sleep Disturbance in Dravet Syndrome: The DREAMS Study. J Clin Sleep Med. 2018; 14(10): 1697-1704.
- Carranza Rojo D, Hamiwka L, McMahon JM, Dibbens LM, Arsov T, Suls A, et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology. 2011 ; 26; 77(4).
- Freilich E, Jones J, Gaillard W, Conry J, Tsuchida T, Reyes C, et al. Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy. Arch Neurol. 2011; 68: 665–71.
- Lim B, Hwang H, Kim H, Chae J, Choi J, Kim K. et al. Epilepsy phenotype associated with a chromosome 2q243 deletion involving SCN1A: Migrating partial seizures of infancy or atypical Dravet syndrome? Epilepsy Res. 2015; 109: 34–9.
- Shein S, Reynolds T, Gedela S, Kochanek P, Bell M. Therapeutic hypothermia for refractory status epilepticus in a child with malignant migrating partial seizures of infancy and SCN1A mutation: a case report. Ther Hypothermia Temp Manag. 2012; 2: 144–9.
- Fang Z, Xie L, Yan L, Lin H, Pan Y, Liu B, et al. Clinical and genetic characteristics of epilepsy of infancy with migrating focal seizures in Chinese children. Epilepsy Res. 2021; 174: 106669.
- Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie H, et al. Gene mutation analysis in 253 Chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS ONE. 2015; 10: e0141782.
- Burgess R, Wang S, Tague A, Boysen K, Yang X, Zeng Q, et al. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann Neurol. 2019; 86: 821–31.
- Brunklaus A, Brünger T, Feng T, Fons C, Lehikoinen A, Panagiotakaki E, et al. The gain of function SCN1A disorder spectrum: novel epilepsy phenotypes and therapeutic implications. Brain. 2022; awac210.
- Jaber D, Gitiaux C, Blesson S, Marguet F, Buard D, Varela Salgado M et al. De novo mutations of SCN1A are responsible for arthrogryposis broadening the SCN1A-related phenotypes. J Med Genet. 2021; 58: 737–742.
- Harkin LA, McMahon JM, Iona X, Dibbens L, Pelekanos JT, Zuberi SM et al. Infantile Epileptic Encephalopathy Referral Consortium: The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007; 130: 843–852.
- Ohashi T, Akasaka N, Kobayashi Y, Magara S, Kawashima H, Matsumoto N, et al. Infantile epileptic encephalopathy with a hyperkinetic movement disorder and hand stereotypies associated with a novel SCN1A mutation. Epileptic Disord. 2014; 16: 208–212.
- Spagnoli C, Frattini D, Rizzi S, Salerno GG, Fusco C. Early infantile SCN1A epileptic encephalopathy: Expanding the genotypephenotype correlations. Seizure. 2019; 65: 62–64.
- Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. 2017; 44: 58-64.
