
Open Access, Volume 10
HLH in a patient with post-surgical Ebstein’s anomaly: Case report
Samuel Mensah1; Bandar Alyami1; Sandeep Banga2; Aaron Shmookler3; Francis L Casey III4; Salwa Gendi4*
1Department of Medicine, West Virginia University, USA.
2Department of Cardiology, Michigan State University, USA.
3School of Medicine, University of Kentucky, USA.
4Department of Pediatrics, West Virginia University, USA.
Salwa Gendi
Department of Pediatrics, West Virginia University, USA.
Email: salwa.gendi@hsc.wvu.edu
Received : Jan 07, 2024,
Accepted : Jan 29, 2024
Published : Jan 31, 2024,
Archived : www.jclinmedcasereports.com
Abstract
This case was interesting as it presents a rare complication in a rare congenital anomaly. We present it to raise awareness about serious fatal complications that could occur in a CHD postoperative patient which may not be related to their surgery. The heart surgery was successful but the patient passed away from complications which arose postoperatively.
A 34-year-old male with Ebstein’s anomaly and severe tricuspid regurgitation status post ASD closure, atrialized ventricular plication and tricuspid annuloplasty at 6 years old presented with symptoms of congestive heart failure. He underwent tricuspid valve replacement and insertion of epicardial dual chamber pacemaker. Patient developed recurrent high fevers on the first night following surgery that were unresponsive to antibiotics and antipyretics. Evaluation revealed hyperferritinemia, thrombocytopenia, anemia and splenomegaly consistent with Hemophagocytic Lymphohistiocytosis (HLH). EVB titers were found to be elevated as a possible inciting infection. He was treated with Dexamethasone, Etoposide and Cyclopsorine, however he developed Heparin Induced Thrombocytopenia (HIT) followed by Disseminated Intravascular Coagulopathy (DIC) and died from a presumed pulmonary embolism.
Conclusion: Surgical intervention outcomes in complex congenital heart disease patients can be complicated by infectious and hematological complications. If more cases of cardiac bypass related HLH are identified, the presence or absence of cold agglutinins as a factor should be studied. Timely recognition will prevent unnecessary mortality and morbidity.
Keywords: HLH; HIT; Ebstein anomaly; Tricuspid valve replacement; PE; Case report.
Abbreviations: EBV: Ebstein Barr Virus; CHB: Congenital Heart Disease; HIT: Heparin Induced Thrombocytopenia; HLD: Hemophagocytic Lymphohistiocytosis.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Gendi S (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Mensah S, Alyami B, Banga S, Shmookler A, Gendi S, et al. HLH in a patient with post-surgical Ebstein’s anomaly: Case report. Open J Clin Med Case Rep. 2024; 2194.
Case Report
Patient was a 34-year-old male with a history of Ebstein’s anomaly, severe tricuspid insufficiency, and atrial septal defect who was s/p ASD closure in 1992 and atrialized ventricular plication, tricuspid annuloplasty and cryoablation in 2001. In 2019, he presented with fatigue, diaphoresis, difficulty breathing, and swelling in his ankles bilaterally.
Echocardiogram (Figures 1,2) and MRI (Figure 3) showed severe dilation of the right atrium, severe tricuspid regurgitation, decreased ventricular systolic dysfunction, and dilated right ventricle. Based on his echocardiogram & MRI, it was determined that he would benefit from a tricuspid valve replacement. Preoperative type and cross was positive for cold agglutinin and needed appropriately matched blood.
Intraoperatively, patient developed ventricular tachycardia and underwent a dual chamber pacemaker placement as well as a tricuspid valve replacement. He was admitted to the ICU intubated and sedated. He initially had severe lactic acidosis with a peak 16.2 mmol/L on the night of surgery and required inotropic support. While intubated and mechanically ventilated the first night after surgery, he had increased work of breathing which improved with nebulized Albuterol and intravenous Solu-medrol.
He had persistent fevers up to 40.1°C and so aggressive noninvasive external cooling with an Arctic Sun device was initiated as well as muscle blockade to alleviate shivering. A chest x-ray was obtained which showed a left lower lobe opacification and Cefepime was initiated for treatment of pneumonia. A Bronchoalveolar Lavage (BAL) was obtained and revealed 4+ PMNs but only 1+ rare gram positive cocci in pairs. He was treated with an Insulin infusion due to diabetes mellitus as well as Heparin for deep venous thrombosis prophylaxis.
Over the following two days, vasoactive infusions were decreased and his lactate decreased went below 2 mmol/L. His repeat echocardiogram showed overall improvement with only minimal tricuspid regurgitation and follow up showed decrease in RA and RV size and normalization of function (Figure 4). He continued to require external cooling however, due to persistent fevers. He also had persistent severe hypoxic respiratory failure secondary to atelectasis that required FIO2 of 100% and inhaled nitric oxide.
On post-operative day 4, a bronchoscopy was performed, which showed thick secretions but normal anatomy. His antibiotics were broadened to Vancomyin and Meropenem and culture then grew Klebsiella.
Despite treatment with broad spectrum antibiotics, patient continued to have persistent fevers. An evaluation for Hemophagocytic Lymphohistiocytosis (HFH) was then initiated and he was found to have an unmeasurably high ferritin greater than 40,000 ng/ml and hypertriglyceridemia at 527 mg/ dl. On exam, he had splenomegaly and hepatomegaly. Laboratory evaluation revealed hepatitis with a peak AST 1621 and ALT of 1384. The hematology team was consulted and Dexamethasone was initiated on their recommendation. A bone marrow aspiration did not reveal hemophagycytosis, leukemia or lymphoma. Soluble IL-2 level was normal at 127 pg/ml. Quantitative NK cell activity was found to be <1%, but qualitative NK cell activity was not obtained. With his combination of persistent fever, splenomegaly, hypertriglyceridemia, hyperferritinemia and cytopenias with anemia and thrombocytopenia, he met the diagnostic criteria for HLH. Treatment was broadened to include Etoposide and Cyclosporine. Despite therapy, he continued to have persistent high fevers. He was then escalated to intrathecal steroids and IVIG treatments per hematology protocol. He had extensive viral testing done, which was positive for EBV IgG and EBNA antibodies as well as a plasma EBV PCR with 1166 IU/ml.
The patient had intermittent neurologic dysfunction throughout his admission. A CT head was unremarkable. EEG revealed diffuse slowing and bursts of rhythmic delta activity consistent with diffuse or multifocal encephalopathy. MRI could not be obtained due to his fresh pacemaker placement. He was able to be successfully extubated and weaned to room air. Following extubation, he was able to communicate intermittently with family appropriately, but continued to have evidence of encephalopathy with slowed responsiveness.
Patient was also found to have bilateral lower extremity DVTs on post-operative day 6 despite treatment despite prophylaxis with intermittent subcutaneous heparin and compression devices. He was started on a Heparin infusion at that time. After being treated with Heparin, his platelets dropped to 58 and he was diagnosed with Heparin-Induced Thrombocytopenia (HIT). His Heparin was changed to Argotroban following return of positive heparin induced antibody via latex Immunoturbidimetric Assay (ILA) followed by serotonin release assay [1]. He had initial improved followed by a worsening of thrombocytopenia and neutropenia. He was diagnosed with HIT induced Disseminated Intravascular Coagulation (DIC). His anticoagulation was then converted to Fondaparinux.
Despite aggressive therapy for HLH, he continued to have high fevers with redeveloped worsened mental status. Following diagnosis of DIC, he was on room air when he had sudden onset of hypoxic respiratory failure with unmeasurably low saturations despite unchanged work of breathing. He then developed severe hypotension followed by cardiac arrest. He was intubated and treated per ALS guidelines including defibrillation but was unable to recover spontaneous circulation. With his known bilateral DVTs and sudden onset of severe hypoxia, a pulmonary embolism with thought to be the most likely cause of death. His family declined an autopsy.
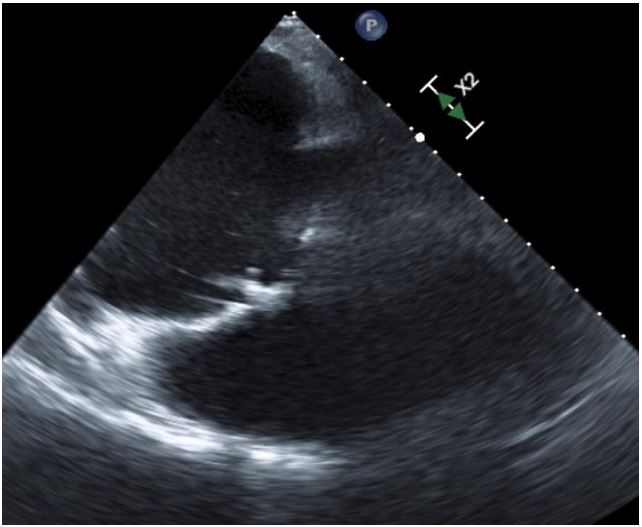
Figure 1: Pre Op TEE showing tricuspid ring, dilated RA
and RV.
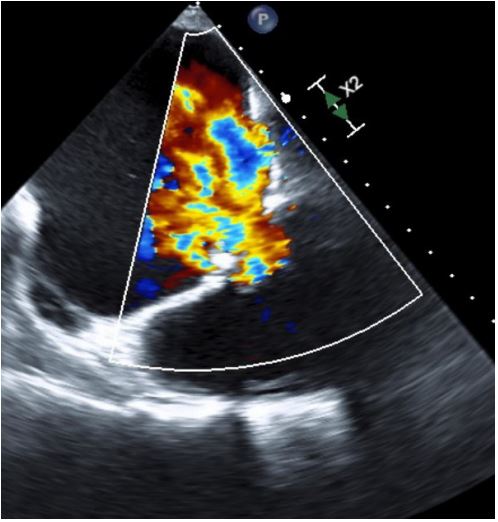
Figure 2: Pre Op TEE with color showing severe Tricuspid Regurgitation (TR).
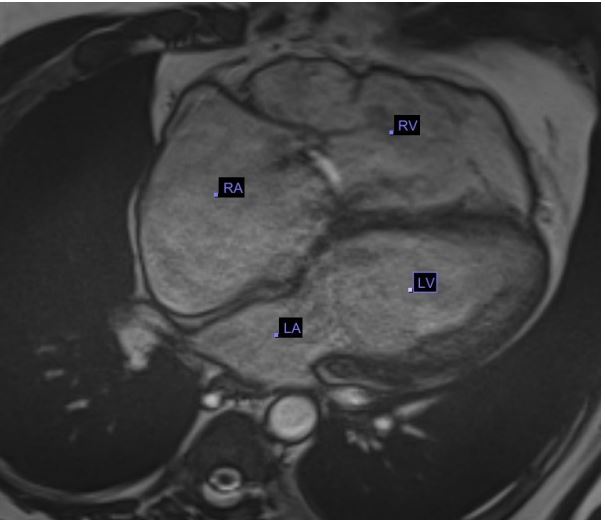
Figure 3: Pre Op MRI showing Axial cine sequence 4
chamber view, showing dilated Right Atrium (RA) & dilated Right Ventricle (RV).
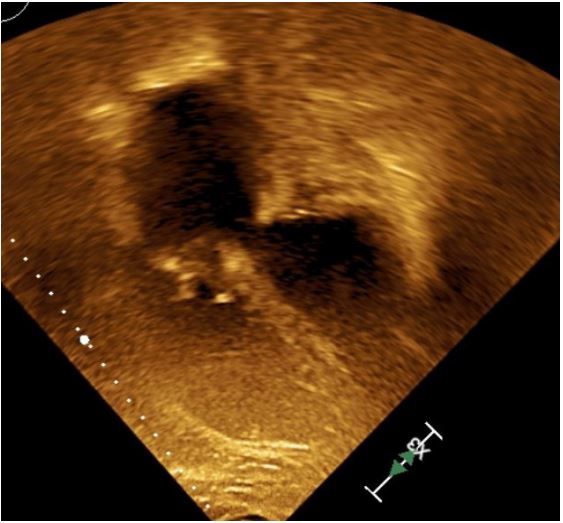
Figure 4: Post OP TTE showing Prosthetic tricuspid
valve, normalization of RA and RV size.
Discussion/Conclusion
Incidence of Ebstein’s anomaly has been reported to be around 1% of all congenital heart diseases [2]. The main defect involves failure of delamination of the tricuspid valve in Ebstein’s anamoly. This results in a number of abnormalities including, adherence and tethering of the leaflets mainly the sepal leaflet of the tricuspid valve to the underlying myocardium and their displacement apically. The anterior leaflet, though attached at the appropriate level of the tricuspid annulus, is larger than normal, and has chordal sail like. The portion of the RV becomes actualized together with dilatation of the left atrium [3]. Approximately 90% of the patients with Ebstein’s anomaly have interatrial communication [4]. There are different surgical approaches for the treatment of Ebstein’s anomaly. Tricuspid valve repair or replacement are the main component as the tricuspid valve defect is the major component of its presentation with plication of atrialized right ventricle, ablation of multiple accessory pathways, bidirectional cavo-pulmonary shunting and specific treatment for right ventricular failure in advanced disease [5].
HLH is classified as either primary which has genetic association or secondary which is acquired [6]. Primary HLH is diagnosed children but secondary HLH can occur at any age group. The secondary HLH has many triggers which include infections, auto-immune diseases and malignancies [6-8]. His Klebsiella pneumonia was also evaluated as a possible trigger for HLH. The BAL was not significantly positive until on day 4 of hospitalization, after initial BAL was negative. Because high fevers began immediately after surgery and four days before pneumonia diagnosed, Klebsiella appears a less likely source.
Another possibility for the trigger for disease was cardiopulmonary bypass. Linthorst reported on the case of a 66-year-old female, who developed HLH following cardiac bypass for aortic valve repair [9]. Similarly, she had increased O2 requirement and persistent fevers following a good cardiac repair. Her condition improved following treatment with Prednisone. Takei also reported on a 50 yo man also with diabetes mellitus, who developed high fevers on day 3 post operative from coronary artery bypass graft surgery [10]. He died of respiratory failure and was found to have HLH on autopsy.
In this case however, HLH seems most likely to be secondary to EBV reactivation. EBV is the most commonly recognized viral trigger of secondary HLH [11]. Serology revealed the patient was negative for EBV IgM, indicating his primary EBV infection was not recent. The elevated EBV PCR with 1166 IU/ml along with the presence of a positive EBV IgG indicates a likely EBV reactivation.
Would the presence of elevated antibodies before surgery be sign of an underlying trigger for development of HLH remains an answered question as the number of patients who develop this complication are unknown and sporadic.
No single test is available but it is multiple findings on specific tests which support the diagnosis as there are multiple patho-physiologies in the causation of the disease process. Elevated serum ferritin and triglycerides are elevated early in the disease. Ferritin elevation is believed to be raised secondary to raised IL1β interleukin. Ferritin levels more than 500 ng/ml well correlate with increased disease activity and hence poor prognosis [12]. There is proliferation of mononuclear phagocyte system in the liver and spleen leading to hepatomegaly and splenomegaly. There is associated cytopenia due to involvement of bone marrow due to hemophagocytosis and depression of hematopoiesis [13]. Plasminogen/factor X activation by cytokines affect platelets leading to bleeding and rarely Disseminated Intravascular Coagulation (DIC) as we saw in our patient [13].
Generally repeated bone marrows aspirations are helpful as we may not see hemophagocytosis initially [14]. Increased expression of α-chain of the soluble interleukin-2 receptor (sCD25) and decreased Natural Killer (NK) cells function are diagnostic [6]. In our patient, we found decreased NK cells at <1%, but cell function was not evaluated. Imaging has no role except we can find hepatosplenomegaly with ascites, thickening of the gallbladder wall, hyperechogenicity and lymphadenopathy of the periportal region [15] on ultrasound, as we have seen in our patient.
Corticosteroid are the mainstay of treatment owing to their cytotoxic effect against lymphocytes and in the inhibition of cytokine expression. N-Dexamethasone is a better choice if the central nervous system is involved since it crosses the blood–brain barrier. Although not clear, but antibiotics and anti-viral drugs may slow down the hemophagocytic process. Cyclosporin and Etoposide have been used due to their role in suppression of T-lymphocyte activity and anti- hemophagocytosis, with monocyte and histiocyte proliferation respectively [16,17]. Despite an initial response, permanent control of the disease can only be achieved by stem-cell transplantation in most cases.
In conclusion, surgical intervention outcomes in complex congenital heart disease patients can be complicated by infectious and hematological complications. As seen in our patient, despite the best possible treatment of the cardiac condition with very good results, the patient died of an apparent pulmonary embolism secondary to HLH and HIT.
If more cases of cardiac bypass related HLH are identified, the presence or absence of cold agglutinins as a factor should be studied.
The diagnosis should be considered in post cardiac surgical patients with unexplained prolonged fever, hepatosplenomegaly and pancytopenia. Timely recognition will prevent unnecessary mortality and morbidity.
References
- Hemos IL HIT-Ab (PF4-H) [Package Insert], Instrumentation Laboratory, Bedford, MA, USA, 10/2016 Revision.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, et al. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly. 2005; 135: 269-81.
- Krieger EV, Valente AM. Diagnosis and management of Ebstein anomaly of the tricuspid valve. Curr Treat Options Cardiovasc Med. 2012; 14: 594-607.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation. 2007; 115: 277-85.
- Stulak JM, Dearani JA, Danielson GK. Surgical management of ebstein’s anomaly. Semin Thorac Cardiovasc Surg Pediatr Card Surg Ann. 2007; 10: 105-11.
- G.E. Janka. Familial and acquired hemophagocytic lymphohistiocytosis Eur J Pediatr, 166 (2007), pp. 95-109.
- J.I. Henter, G. Elinder, A. Ost Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society Semin Oncol. 1991; 18: 29-33.
- AH Filipovich. Hemophagocytic lymphohistiocytosis (HLH) and related disorders Hematology Am Soc Hematol Educ Program. 2009; 127-131.
- L. Linthorst, Unexpected hemophagocytic syndrome in a post-cardiac surgery patient. Critical Care. 2011; 15: 440.
- K Takei. Marchiafava-Bignami disease with haemophagocytic lymphohistiocytosis as a postoperative complication of cardiac surgery. BMJ Case Reports CP 2019; 12: e230368.
- NG Rouphael. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007; 7(12): 814-22.
- R Dhote, J Simon, T Papo, et al. Reactive hemophagocytic syndrome in adult systemic disease: Report of twenty-six cases and literature review Arthritis Rheum. 2003; 49: 633-639.
- Creput L, Galicier S, Buyse, et al. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med. 2008; 34: 1177-1187.
- JW Farquhar, AE Claireaux. Familial haemophagocytic reticulosis Arch Dis Child. 1952; 27: 519-525.
- F Chateil, M Brun, Y Perel, et al. Abdominal ultrasound findings in children with hemophagocytic lymphohistiocytosis Eur Radiol. 1999; 9: 474-477.
- S. Imashuku, K. Kuriyama, T. Teramura, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis J Clin Oncol. 2001; 19: 2665-2673.
- G.E. Janka Hemophagocytic syndromes. Blood Rev. 2007; 21: 245-253.
