
Open Access, Volume 10
A case report: Paracellin-1 gene mutation and nephrocalcinosis
Ilkin Hamid-Zada*; Ozgur Efiloglu; Bekir Demirtas; Asif Yildirim
Department of Urology, Istanbul Medeniyet Univercity, Istanbul/Turkey.
Ilkin Hamid-Zada
Department of Urology, Istanbul Medeniyet Univercity, Istanbul/Turkey.
Tel: +905388466687;
Email: ilkin081994@gmail.com
Received : Dec 29, 2023,
Accepted : Jan 22, 2024
Published : Jan 31, 2024,
Archived : www.jclinmedcasereports.com
Abstract
Nephrocalcinosis is quite common in society. Bilateral kidney stones, family history of stones should be examined with genetic testing. Here we present our patient who was previously diagnosed with a genetic mutation and had a bilateral kidney stones. Renal function tests and electrolytes should be evaluated carefully. It is very important to follow up closely
Keywords: Paracellin-1 gene; Claudin-16 protein; Nephrocalcinosis; Consanguineous marriage; Hypomagnesemia.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Hamid-Zada I (2024)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Hamid-Zada I, Efiloglu O, Demirtas B, Yildirim A. A case report: Paracellin-1 gene mutation and nephrocalcinosis. Open J Clin Med Case Rep. 2024; 2190.
Introduction
Nephrolithiasis are highly prevalent and affecting approximately 10% of adults worldwide, incidence of stone disease is increasing and nephrolithiasis is a major health problem worldwide. About 80% of urinary stones are composed of calcium and hypercalciuria is found in up to 40% of stone-formers [1]. A genetic control of hypercalciuria has been suspected for a long time. Approximately half of the patients with idiopathic hypercalciuria have a family history of nephrolithiasis, and stone disease is more common seen a family member [1]. Claudin-16 is a renal tight junction protein encoded by the Claudin-16 gene [2]. As we know, it is a rare autosomal recessive disease. Claudin-16 gene plays a role in the absorption of magnesium and calcium in the thick ascending limb of the loop of Henle [2]. It is characterized by hypercalciuria and hypomagnesemia. We report an extremely rare case of Claudin-16 gen mutation patient kidney stones.
Case Report
A 38-year-old female patient presented with intermittent bilateral flank pain. She was a first child as a result of consanguineous marriage. She has a hyperparathyroidism and ventricular septum defect. She is followed by nephrology department and cardiology department due to ventral septal defect. She has a previously undergone operation four time left-side percutaneous nephrolithotomy operations, bilateral ureterorenoscopy also bilateral retrograde intrarenal surgery operations. In the Dimercapto Succinic Acid (DMSA) taken on the patient, it was seen that the left kidney was functioning at 39% and the right kidney was functioning at 61%. The patient’s magnesium values were below the normal limit value, and the creatine value was 1.2 mg/dL on average (Figure 2). Other laboratory investigations serum creatinine 1,23 mg/dL, serum urea 31 mg/dL, serum uric acid 6,1 mg/dL, sodium 143 mmol/L, potassium 4,5 mmol/L, calcium 9,2 mg/dL, phosphorus 1,3 mg/dL (below normal range). The results of 24-h urine tests were as follows: magnesium 121,31 mg/day, calcium 263,57 mg/day, oxalate 115,06 mg/day (normal range 4-31), citrate 162,68 mg/day (normal range 320-1260), uric acid 365,8 mg/day. The patient had previously used Potassium Citrate and Thiazide group drugs. Computer tomography taken at her last admission showed multiple stones filling the bilateral kidneys (Figure 1).
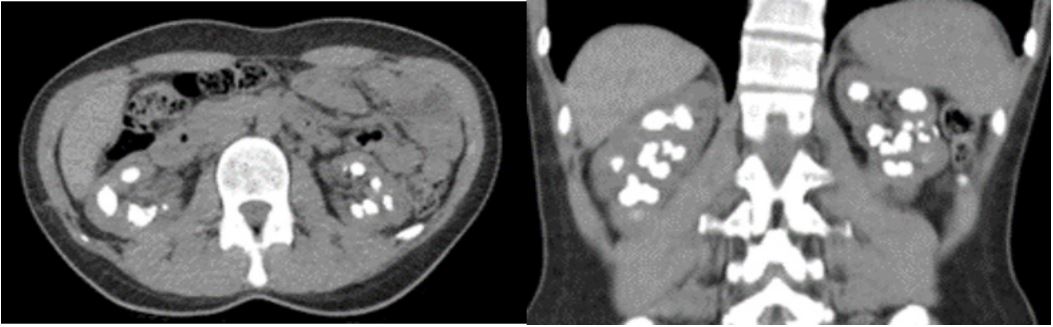
Figure 1: CT show a bilateral kidney stones.
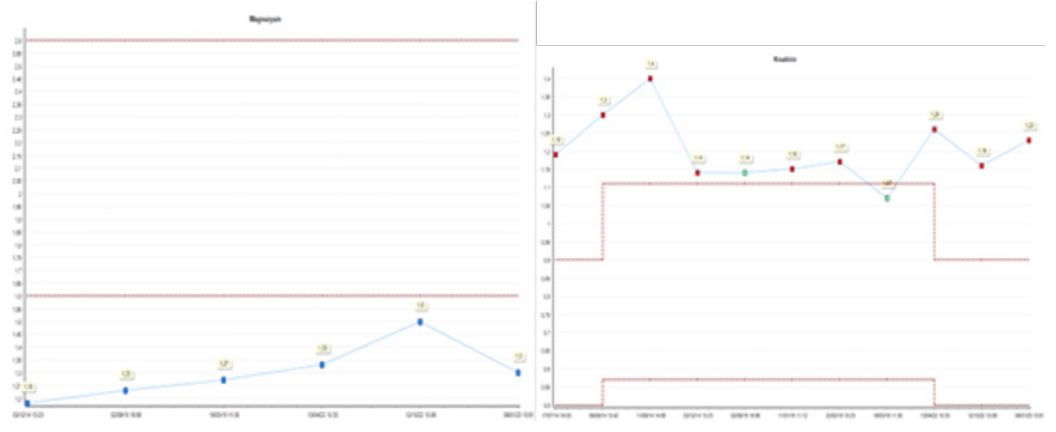
Figure 2: Magnessium and creatinine value.
Discussion
Firstly, familial primary hypomagnesemia with hypercalciuria and nephrocalcinosis clinical description by Michelis et al. in 1972, and nowadays reported approximately 120 cases worldwide [3]. Familial Primary Hypomagnesemia with Hypercalciuria and Nephrocalcinosis (FHHNC) is a hereditary autosomal recessive disorder, also related to mutations in paracellin-1 gen mutation. This protein located in the thick ascending limb of Henle [4]. Also we know that, paracellular protein is a responsible for a magnesium and calcium reabsorption [5]. Also female patient affected more than a male patient [5].Therefore, discuss about relationship between estregen level and nephrocalcinosis [5]. The most common finding nephrocalcinosis due to calcium deposit and kidney cysts [6]. Also we can see ocular defects especially Claudin-19 gene mutation [6]. A rare related to Familial Primary Hypomagnesemia with Hypercalciuria and Nephrocalcinosis (FHHNC) mutation between chondrocalcinosis [6]. Here we report a female patient with bilateral nephrocalcinosis, hypomagnesemia, hypophosphatemia and hypocitraturia. The patient was diagnosed with paracellin gene mutation 30 years ago. Nephrocalcinosis and recurrent infection play a important role a chronic kidney disease due to paracellin-1 gene mutation. Also, paracellin-1 gene mutation affect for a renal function is a unclear. Konrad et al. had a related phenotype-genotype correlation between renal impairment progression in FHHNC patients [3]. This study suggested that the presence of at least one partial-function Claudin-16 gene allele is sufficient to predict the clinical phenotype. It is also seen in distal renal tubular acidosis in FHHNC patients. Weber S et al. found that 17 of 20 patients with Claudin-16 gene mutation had distal renal tubuler asidosis [3]. The reason for this is due to distal acidification defect. Although it is controversial in the literature but most of patient especially in the 2nd or 3rd decade of life Familial Primary Hypomagnesemia with Hypercalciuria and Nephrocalcinosis (FHHNC) progresses to endstage renal failure [7]. Blanchard et al. has been shown that consanguineous marriage is quite common in this mutation group. This causes family members tend to be hypercalciuria and hypomagnesia, as in our patient [8]. There is no specific method for the treatment of this disease. Paracellin-1 gen mutation patients who have a nephrocalcinosis can be treated by thiazide diuretics (reduce urinary calcium excretion), potassium citrate, Magnesium supplement. İn addition salt restriction and high fluid intake may be helpful. The only treatment of end-stage renal disease is renal replacement therapy [9].
Conclusion
Finally, metabolic evaluation should be performed in high-risk patients with a family history of stones, bilateral kidney stones, and frequently recurring stones. Supportive therapy including magnesium salts, thiazides, and citrate, as well as abundant fluid intake and dietary restrictions, are widely used to delay the progression of renal failure. Kidney transplantation remains the only way to curative option for kidney failure in these patients. In this group of patients who have a Paracellin - 1 gene mutation, calcium/creatine, magnesium/creatine ratios should be followed and they should be followed frequently and approached more carefully in order to prevent rapid development of renal failure. We recommend a genetic evaluation in young patients who have a nephrocalcinosis and a consanguineous marriages.
References
- Devuyst O, Pirson Y. Genetics of hypercalciuric stone forming diseases. Kidney International. 2007; 72: 1065-72.
- Türkmen M, Kasap B, Soylu A, Böber E, Konrad M, et al. Paracellin-1 gene mutation with multiple congenital abnormalities. Pediatric Nephrology. 2006; 21(11): 1776-8.
- Lu J, Zhao X, Paiardini A, Lang Y, Bottillo I, et al. Familial hypomagnesaemia, Hypercalciuria and Nephrocalcinosis associated with a novel mutation of the highly conserved leucine residue 116 of Claudin 16 in a Chinese patient with a delayed diagnosis: A case report. BMC Nephrol. 2018; 19(1).
- Enríquez R, Sirvent AE, Amorós F, Martínez M, Cabezuelo JB, et al. Renal Hypomagnesemia, Hypercalciuria and Nephrocalcinosis in a Middle-aged Man. 2003; 37(1): 93-5.
- Wolf MTF, Dötsch J, Konrad M, Böswald M, Rascher W. Follow-up of five patients with FHHNC due to mutations in the Paracellin-1 gene. Pediatric Nephrology. 2002; 17(8): 602-8.
- Vall-Palomar M, Madariaga L, Ariceta G. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Pediatric Nephrology. 2021; 36(10): 3045-55.
- Al-Haggar M, Bakr A, Tajima T, Fujieda K, Hammad A, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Unusual clinical associations and novel claudin16 mutation in an egyptian family. Clin Exp Nephrol. 2009; 13(4): 288-94.
- Türkmen M, Kasap B, Soylu A, Böber E, Konrad M, et al. Paracellin-1 gene mutation with multiple congenital abnormalities. Pediatric Nephrology. 2006; 21(11): 1776-8.
- Prot-Bertoye C, Houillier P. Claudins in renal physiology and pathology. Genes. MDPI AG. 2020; 11.
