
Open Access, Volume 9
Cutaneous lesions, polyuria, polydipsia, and adenopathies in a pediatric patient
Lucía García Sirvent, MD; Leticia Beas Porcel, MD; Joaquín Espiñeira Sicre, MD; Juan Ruiz Sánchez, MD; Joan García Vilar, MD; Laura Garcia-Fernandez, PhD1
Department of Dermatology, Hospital Universitario Sant Juan, Alicante, Spain.
Lucía Garcia Sirvent
N-332, s/n, 03550 Sant Joan d’Alacant, Alicante, Spain.
Email: luciagarciasirvent@gmail.com
ORCID ID: 0000-0003-3740-5485
Received : Oct 10, 2023,
Accepted : Nov 07, 2023
Published : Nov 10, 2023,
Archived : www.jclinmedcasereports.com
Abstract
This abstract presents a case of multisystemic Langerhans cell histiocytosis (LCH) in a 3-year-old boy with diverse clinical manifestations, including language delay, recurrent acute otitis media, pruritic scalp plaques, and erythematous trunk papules. Laboratory and radiological findings supported the diagnosis, which was confirmed by histopathological analysis and immunohistochemical markers. The case underscores the importance of recognizing skin involvement as an initial presentation and highlights evolving therapeutic strategies, including targeted inhibitors, for improved LCH management.
Keywords: Histiocytosis; Langerhans cells; BRAFV600E.
Abbreviations: LCH: Langerhans cell histiocytosis; MAPK kinase: mitogen-activated protein kinase; BRAF: v-raf murine sarcoma viral oncogene homolog B1; MEK: methyl ethyl ketone.
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Sirvent LG (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Sirvent LG, Porcel LB, Sicre JE, Sánchez JR, Vilar JG, Garcia-Fernandez L. Cutaneous lesions, polyuria, polydipsia, and adenopathies in a pediatric patient. Open J Clin Med Case Rep. 2023; 2153.
Case Presentation
A 3-year-old boy with a significant language delay and a history of repeated acute otitis media presented with pruritic, yellowish-crusted plaques on the scalp and erythematous excoriated papules on the trunk (Figure 1). Concurrently, the patient experienced polyuria, polydipsia (up to six liters per day), anorexia, and asthenia. Physical examination revealed cervical and inguinal adenopathies. Blood and urine tests showed increased plasma osmolality, hypernatremia, hyperkalemia, and hyperchloremia, with low urine density, osmolarity, and urinary sodium.
Skeletal survey was performed, finding radiolucent images corresponding to osteolytic lesions in the skull, pelvis and right femur (Figure 2). Brain MRI showed thickening of the pituitary stalk with total absence of neurohypophysis.
Histopathological findings: A skin biopsy displayed a dense histiocytic infiltrate in the papillary dermis, accompanied by a mixed inflammatory infiltrate and abundant eosinophils (Figure 3). Immunohistochemistry confirmed the presence of CD68, CD1a, S100, and Langerin (CD207), supporting the diagnosis of LCH. Additionally, the BRAFV600E mutation study yielded positive results.
Diagnosis
The patient was diagnosed with multisystemic langerhans cell histiocytosis with bone, skin, lymph node, and pituitary gland involvement (central diabetes insipidus).
Background, discussion and conclusions
Langerhans cell histiocytosis (LCH) is an inflammatory neoplasm derived from myeloid precursors and caused by mutations in the mitogen-activated protein kinase (MAPK kinase) pathway, the most frequent being the BRAFV600E mutation. This results in an excessive proliferation and accumulation of langerhans cells in various organs, leading to diverse clinical manifestations [1].
The incidence of LCH is considered very low, with a prevalence of 1 to 5 cases per million population under 15 years of age. Historically, LCH has been classified into syndromic subtypes based on clinical manifestations, including Letterer-Siwe disease, Hand-Schüller-Christian disease, eosinophilic granuloma, and Hashimoto-Pritzker disease. However, a more contemporary approach to classification has emerged following the recommendations of the European Euro Histio Net consortium in 2013 [2].
Currently, the preferred classification system for LCH is based on whether there is single organ or multiorgan involvement. This approach takes into account the extent of organ systems affected and provides a more comprehensive understanding of the disease’s presentation and prognosis.
From a clinical perspective, LCH is a multi-systemic disorder affecting various organ systems, including the bones, skin, pituitary gland, liver, bone marrow, spleen, lymph nodes, and lungs. Of particular significance is the involvement of the skin, which serves as the primary presenting manifestation in a substantial proportion of cases, approximately 80%. Additionally, skin involvement acts as a valuable indicator of concurrent multi-organ participation, with a notable majority of patients who present to dermatologists with cutaneous manifestations (ranging from 87% to 93%) demonstrating the presence of LCH in other organ systems.
The dermatological features of LCH encompass a diverse range of manifestations, including seborrheic dermatitis-type rash, erythematous/brownish papules, eczematous lesions, fold dermatitis, hypopigmented macules, purpura, and petechiae [1,3]. Less frequent cutaneous presentations have also been documented, such as papulo-pustular rash, erythematous-violaceous nodules, and the distinctive appearance known as «blueberry muffin baby” [1,3].
The diagnosis is confirmed histologically by the presence of langerhans cell infiltrates in the affected tissues, along with positive immunohistochemistry for CD1a and Langerin [4].
Treatment options for LCH vary depending on the extent of involvement and may involve topical corticosteroids for localized cutaneous forms and oral corticosteroids with cytostatic drugs for multisystemic forms. Additional therapeutic strategies, including targeted inhibitors (BRAF and MEK inhibitors), are currently under investigation.
Learning points: To highlight the importance of the skin as a guiding sign for diagnosis of langerhans cell histiocytosis, as well as an indicator of multisystem involvement.
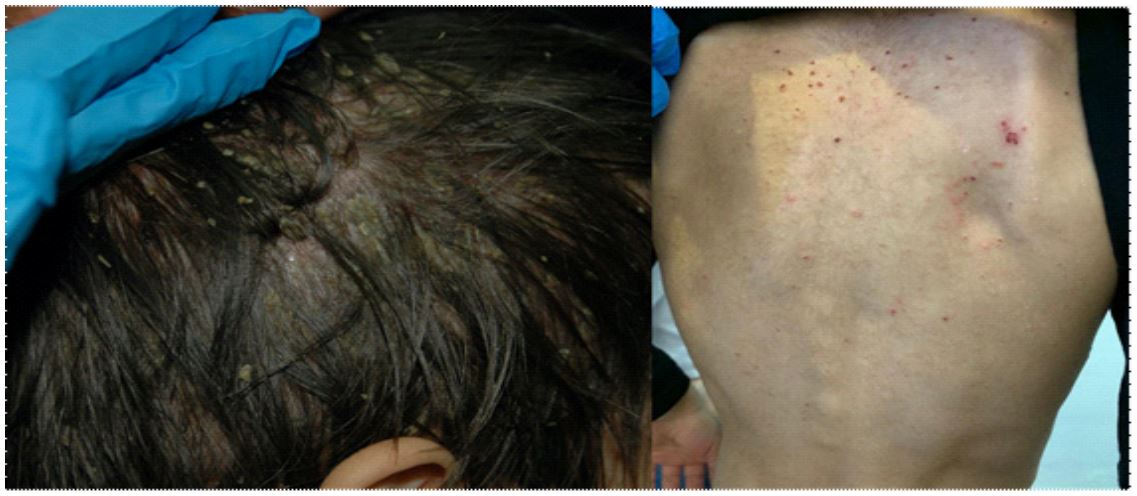
Figure 1: Scaly plaques on the scalp with a seborrheic appearance. Erythematous-orange papules and excoriation lesions on the trunk.
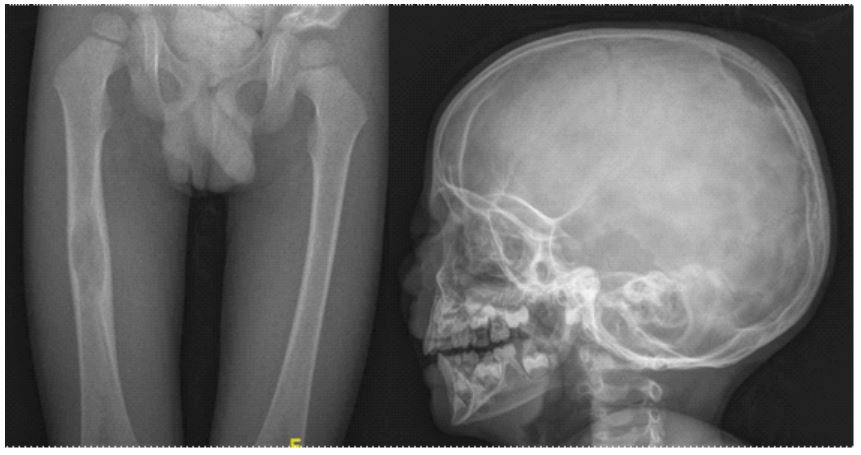
Figure 2: Bone radiographs showing radiolucent lesions in the right femoral
diaphysis and skull.
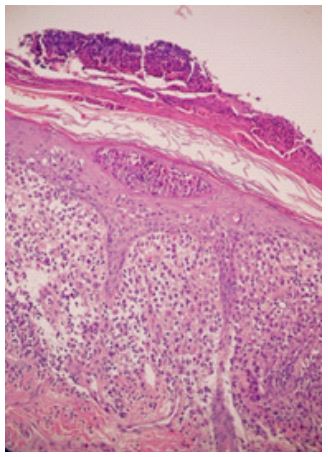
Figure 3: Skin biopsy stained
with hematoxylin-eosin, demonstrating a dense cellular infiltrate in the papillary dermis,
composed of histiocytes and
mixed inflammatory cells.
Declarations
Funding sources: None.
Conflicts of interest: None declared.
Patient consent: We declare that the patient gave written consent for his medical information to be published in print and online with the understanding that this information may be publicly available.
References
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: History, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78(6):1035–44.
- Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years: Guidelines for Langerhans Cell Histiocytosis. Pediatr Blood Cancer. 2013;60(2):175–84.
- Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: Diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78(6):1047–56.
- Fraitag S, Emile J-F. Cutaneous histiocytoses in children. Histopathology. 2022;80(1):196–215.
- Donadieu J, Larabi IA, Tardieu M, Visser J, Hutter C, Sieni E, et al. Vemurafenib for refractory multisystem Langerhans cell histiocytosis in children: An international observational study. J Clin Oncol. 2019;37(31):2857–65.
