
Open Access, Volume 9
Neuro-Ophthalmologic Igg4-Related disease
Tribovane Denil; Siurana-Montilva Sahyly*
Hospital Universitario Vall d´Hebrón, Spain.
Siurana-Montilva Sahyly
Hospital Universitario Vall d´Hebrón, Spain.
Email: sahy.siurana.idi@gencat.cat
Received : Aug 01, 2023,
Accepted : Oct 23, 2023
Published : Oct 27, 2023,
Archived : www.jclinmedcasereports.com
Abstract
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Siurana-Montilva S (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Denil T, Siurana-Montilva S. Neuro-Ophthalmologic Igg4-Related disease. Open J Clin Med Case Rep. 2023; 2141.
Introduction
IgG4-Related Disease (IgG4-RD) is a multiorgan, immune-mediated, fibro-inflammatory disease of unknown etiology that has integrated a spectrum of conditions previously not recognized as related to each other or any systemic disease. It has an estimated prevalence of 4.6 per 100,000, however, the exact prevalence of IgG4-RD is difficult to estimate since there have been few published studies, and these studies used different criteria for diagnosis, tended to enroll small groups of patients with heterogeneous pathology, and had uncertain generalizability [1,2].
Histology shows a very characteristic triad of infiltration of IgG4-bearing plasmocytes, storiform fibrosis and obliterative phlebitis, which can be accompanied by high IgG4 levels in the serum [3]. However, much remains unknown about the behavior of IgG4 in vivo, the participation of this molecule in disease, and whether its role in IgG4-related disease is primary or secondary [4].
Ophthalmic manifestations of IgG4-RD are common, affecting approximately a quarter of the IgG4-RD patients, and involvement has been reported in nearly every orbital structure [5]. Neurologic manifestations of IgG4-RD are rare, with the most frequent being secondary to either hypertrophic pachymeningitis or hypophysitisb [6].
Case Presentation
A 58-year-old man with a medical history of hypertension, controlled with candesartan and hydrochlorothiazide, and surgical history of left eye cataract surgery using retrobulbar anesthesia 1 month prior presented with complaints of blurry vision and headaches since the surgery. Ophthalmologic examination revealed left proptosis, chemosis and limitation of upwards and downwards eye gaze, without diplopia. A central scotoma and hyporeactive left pupil were also documented without clear relative afferent pupillary defect. Routine laboratory results were unremarkable. Urinalysis did not show proteinuria or hematuria. Computer Tomography (CT) (Figure 1) and Magnetic Resonance Imaging (MRI) (Figure 2) were performed that revealed a left intraorbital infiltrating soft tissue lesion with involvement of the superior and lateral rectus muscles and extension into the preseptal space, involving and enlarging the lacrimal gland. The lesion displaced the left optic nerve medially and the globe inferiorly. A right infiltrating orbital apex lesion with similar imaging characteristics was also documented. Intracranially, temporal and tentorial pachymeningeal thickening was also noted.
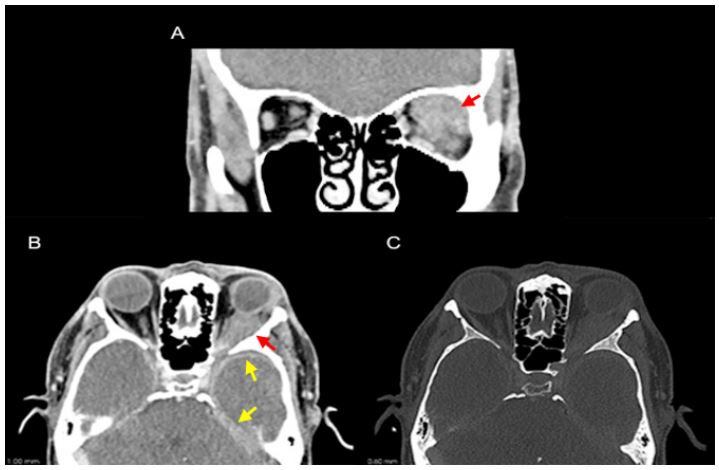
Figure 1: CT scan demonstrating a soft tissue density mass in the left orbit infiltrating the post-septal intra and extra-conal
spaces in the left superior quadrant (A and B; red arrow) with antero-lateral extension to the left lacrimal gland and posterior extension to the cavernous sinus. Intracranially, bilateral left predominant temporal and tentorial pachymeningeal can
also be noted (B; yellow arrow). No bony destruction was documented (C).
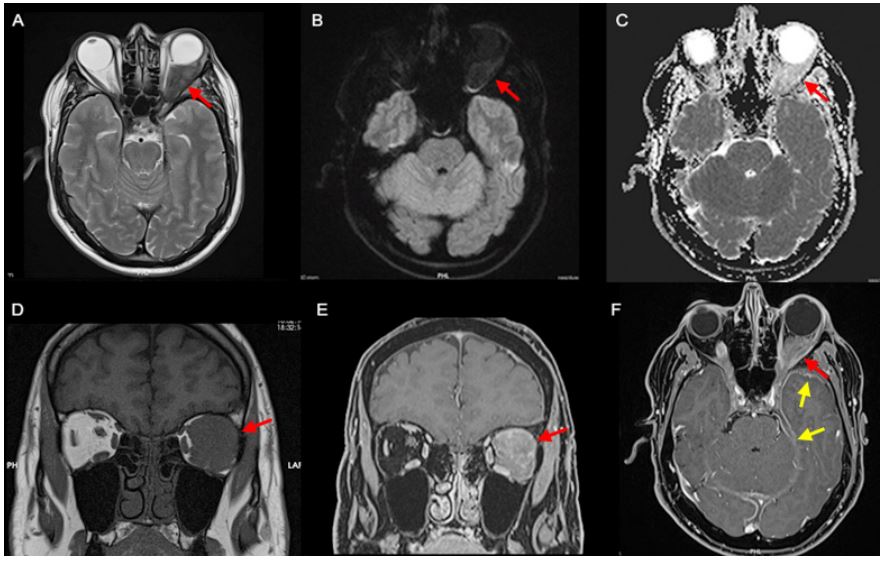
Figure 2: MRI depicting a left orbital retrobulbar mass hypointense on T2-weighted imaging due to its fibrous nature (A), without restriction on diffusion-weighted imaging (B and C) and spontaneously hypointense on T1-weighted imaging (D) with
strong homogeneous enhancement depicted on the fat saturated post-contrast T1-weighted images (E and F) (red arrow).
The lesion involved the left lateral and superior rectus muscles, with anterolateral extension to the ipsilateral lacrimal gland
and posterior extension to the cavernous sinus. A smaller soft tissue enhancing lesion was also documented in the right orbital apex with extension to the right cavernous sinus, with the same signal characteristics and, intracranially, left-predominant
bilateral temporal and tentorial pachymeningeal thickening was better depicted (F; yellow arrow).
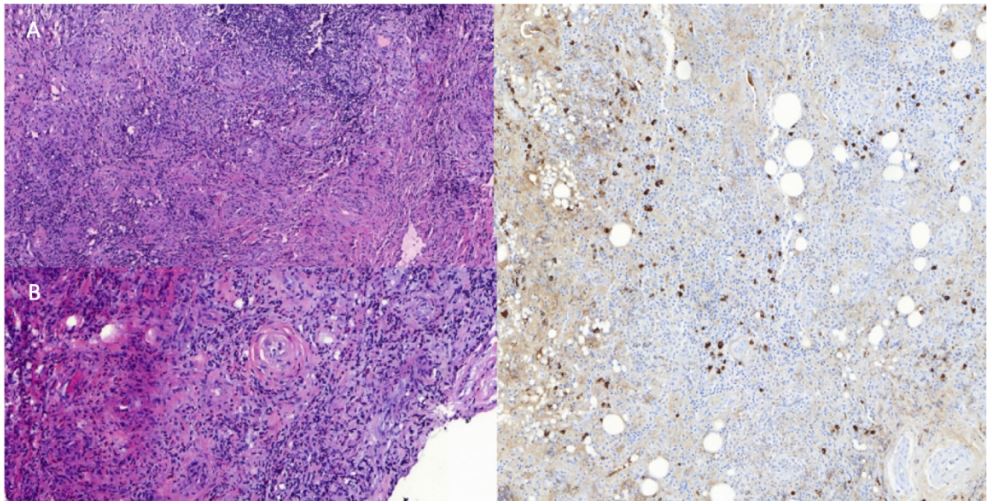
Figure 3: Histopathological analysis with hematoxylin and eosin stain revealed a rich lymphoplasmocytic infiltrate in a
background of fibrosis with a slightly whorled pattern (A) with signs of periphlebitis and obliteration of the vascular lumen
(B). Imunohistochemistry depicted a rich infiltrate of IgG4+ cells (64 IgG4+ cells/HPF and a ratio of IgG4/IgG of 44,4%) (C).
An orbital biopsy was performed which excluded malignant disease. Histopathological analysis revealed elevated counts of 64 IgG4+ cells/HPF and a ratio of IgG4/IgG of 44, 4%. Fibrous changes with a slight whorled pattern was also observed in the tissue specimen (Figure 3). Serum IgG4 analysis was then obtained and found to be elevated at 137 mg/dL.
The diagnosis of IgG4-Related Disease (IgG4-RD) was proposed and the patient started an oral prednisone therapy regimen (induction at 60 mg per day, followed by a maintenance period of 30 mg per day during 5 months and slow taper afterward). Given the concern for subclinical neuronal injury, the patient initiated rituximab right from the start with two doses of 1 g intravenously separated by 2 weeks. Follow-up clinical examination after 3 months showed improvement in visual acuity, proptosis and chemosis.
Discussion
The pathophysiology of IgG4-RD remains incompletely understood. It is believed to involve autoimmune mechanisms driven by dysregulated immune responses. The precise triggers for immune dysregulation and its ability to induce tissue fibrosis are unknown, but both genetic and environmental factors may play a role [4,7].
IgG4-RD usually presents in a subacute fashion. In many cases, symptoms and evidence of organ dysfunction may be present for months or years before the diagnosis is established [6]. There can be periods of stability or less frequently, spontaneous improvement in one organ before re-emergence in another [8].
The most typical presentation of IgG4-Related Orbital Disease (IgG4-ROD) involves subacute progressive, painless unilateral or bilateral periorbital or lacrimal gland swelling. If left untreated, progression to proptosis may ensue, which results from infiltration of the inflammation into the extraocular muscles, orbital fat, or connective tissue [8]. Although there is a predilection for the muscle and tendon of the lateral rectus muscle (contrasting with involvement of the muscle belly and preferential involvement of the inferior rectus in thyroid eye disease), any of the extraocular muscles can be affected with the potential for muscle enlargement and restriction of motility resulting in diplopia [9].
An increased risk of lymphoproliferative disorders, particularly Mucosa-Associated Lymphoid Tissue (MALT) lymphoma, has been suggested in IgG4-RD and this entity has to be excluded as the clinical course and symptoms of the disease can be similar [10].
On MRI, the orbital masses in IgG4-ROD frequently demonstrate low signal on T1 and T2-weighted images due to the presence of fibrosis, with avid enhancement in T1 post-gadolinium sequences [11]. Infra-orbital nerve enlargement has been shown to correlate well with IgG4-ROD [12]. Although some patients with trigeminal nerve involvement report facial paresthesia, many are asymptomatic probably because involvement is limited to the epineurium [13]. Our case did not have imaging findings suggestive of infraorbital nerve involvement.
Patients with IgG4-RD-associated hypertrophic pachymeningitis are more frequently men, with a peak incidence in the sixth and seventh decades of life [14]. Meningeal involvement may be contiguous with disease in the orbit or pituitary gland and these patients may show elevations of cerebrospinal fluid IgG4 level, indicating intrathecal production of IgG4. On MRI, the dura may show smooth thickening with homogeneous diffuse linear enhancement (as in the case presented), or focal nodular thickening, mimicking a meningioma, and may result in compression of surrounding structures, especially cranial nerves [15].
Patients with IgG4-related infundibulo-hypophysitis present with visual symptoms from mass effect or endocrine dysfunction in the setting of a pituitary mass or a thickened pituitary stalk [16]. Radiologic studies demonstrate an enlarged pituitary gland with thickened stalk and MRI can show an absence of the regular precontrast T1 hyperintensity of the posterior pituitary gland, which can be associated with central diabetes insipidus [17].
Clearly defined diagnostic criteria remain lacking, with consensus guidelines recommending that clinical, radiographic, serological and pathological evidence be taken together, as no finding is individually diagnostic.
The American College of Rheumatology/European Alliance of Associations For Rheumatology (ACR/ EULAR) classification criteria divides IgG4-RD into four different groups according to different phenotypes: pancreato-hepatobiliary disease (31%) retroperitoneal fibrosis with or without aortitis (24%) head and neck-limited disease (24%) and classic Mikulicz’s syndrome with systemic involvement (22%) [18]. According to these criteria, our patient falls under the head and neck-limited disease group because of the orbital disease as meningeal involvement is not yet included as a part of this classification.
The revised comprehensive diagnostic criteria for IgG4-RD proposed by the Japanese IgG4 team defines three items for the diagnosis of IgG4-RD: Diffuse or localized swelling or masses in one or more organs; Serum IgG4 levels greater than 135 mg/dl; and a histopathologic examination of the affected organs revealing dense lymphoplasmacytic infiltration, storiform fibrosis, and/or obliterative phlebitis along with elevated IgG4-positive plasma cells (>10-50 per high-power field depending on the organ or ratio of IgG4+/ IgG+ of >40%) [19]. Our case fulfills all three criteria corresponding to a definite diagnosis of IgG4-RD.
The first line of treatment is oral prednisone. However, since there is a risk of subclinical neuronal injury, current recommendations mandate initiation of rituximab from the beginning (2 doses of 1 g intravenously, separated by 2 weeks and repeated at 6-month intervals) with follow-up neuroimaging at 3 months [20,21].
Conclusion
In conclusion, imaging plays a key role in avoiding treatment delays as prompt recognition of IgG4- RD is important not only because it is generally highly treatable, but also because its treatment frequently differs substantially from that of other conditions that present in a similar fashion, such as lymphoma, and performing a directed biopsy is fundamental as histopathological examination remains the main way to definitively diagnose IgG4-RD.
References
- Saitakis G, Chwalisz BK. The neurology of IGG4-related disease. J Neurol Sci. 2021; 424: 117420.
- Aryasit O, Tiraset N, Preechawai P, Kayasut K, Sanghan N, et al. IgG4-related disease in patients with idiopathic orbital inflammation. BMC Ophthalmol. 2021; 21: 356.
- Mulay K, Aggarwal E, Honavar SG. Clinicopathologic features of orbital immunoglobulin G4-related disease (IgG4-RD): a case series and literature review. Graefes Arch Clin Exp Ophthalmol. 2015; 253: 803-9.
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012; 366: 539-51.
- Kashii S. IgG4-related disease: A neuro-ophthalmological perspective. J Neuroophthalmol. 2014; 34: 400-7.
- Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM. Neurological Manifestations of IgG4-Related Disease. Curr Treat Options Neurol. 2017; 19: 14.
- Maehara T, Moriyama M, Nakamura S. Pathogenesis of IgG4-related disease: A critical review. Odontology. 2019; 107: 127-132.
- Chwalisz BK, Stone JH. Neuro-ophthalmic complications of IgG4-related disease. Curr Opin Ophthalmol. 2018; 29: 485-494.
- Pitz S. IgG4-assoziierte Erkrankung [IgG4-related disease]. Ophthalmologe. German. 2021; 118: 787-793.
- Chen LYC, Mattman A, Seidman MA, Carruthers MN. IgG4-related disease: What a hematologist needs to know. Haematologica. 2019; 104: 444-455.
- Kurowecki D, Patlas MN, Haider EA, Alabousi A. Cross-sectional pictorial review of IgG4-related disease. Br J Radiol. 2019; 92: 20190448.
- Ohshima K, Sogabe Y, Sato Y. The usefulness of infraorbital nerve enlargement on MRI imaging in clinical diagnosis of IgG4-related orbital disease. Jpn J Ophthalmol. 2012; 56: 380-2.
- Sogabe Y, Miyatani K, Goto R, Ishii G, Ohshima K, et al. Pathological findings of infraorbital nerve enlargement in IgG4-related ophthalmic disease. Jpn J Ophthalmol. 2012; 56: 511-4.
- Lu LX, Della-Torre E, Stone JH, Clark SW. IgG4-related hypertrophic pachymeningitis: Clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014; 71: 785-93.
- Sapkota B, Rampure R, Gokden M, Kanuru S. IgG4-Related Disease Presenting as Hypertrophic Pachymeningitis. Cureus. 2022; 14: e21850.
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: A new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011; 96: 1971-80.
- Decker L, Crawford AM, Lorenzo G, Stippler M, Konstantinov KN, et al. IgG4-Related Hypophysitis: Case Report and Literature Review. Cureus. 2016; 8: e907.
- Wallace ZS, Zhang Y, Perugino CA, Naden R, Choi HK, et al. ACR/EULAR IgG4-RD Classification Criteria Committee. Clinical phenotypes of IgG4-related disease: An analysis of two international cross-sectional cohorts. Ann Rheum Dis. 2019; 78: 406-412.
- Umehara H, Okazaki K, Kawa S, et al. The 2020 Revised Comprehensive Diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021; 31: 529-533.
- AbdelRazek MA, Venna N, Stone JH. IgG4-related disease of the central and peripheral nervous systems. Lancet Neurol. 2018; 17: 183-192.
- Maritati F, Peyronel F, Vaglio A. IgG4-related disease: a clinical perspective. Rheumatology (Oxford). 2020; 59: iii123-iii131.
