
Open Access, Volume 9
Kidney injury in COVID-19 patients: Clinical manifestations and pathogenesis
Xie Na1; Zhiwu Lu2; Zibiernisa Yaku2; Qianqian Qian2; Yue Li1; Xiao Liu1; Siqi Wu1,3; Zhanjun Shu1,4*
1Infectious Diseases and Nephropathy, The Fourth Clinical College of Xinjiang Medical University, Urumqi 830000, China.
2Infectious Diseases and Nephropathy, The Eighth Affiliated Hospital of Xinjiang Medical University, Urumqi 830000, China.
3Traditional Chinese Medicine, Xinjiang Urumqi Hospital of Traditional Chinese Medicine, Urumqi 830000, China.
4National Clinical Research Base of Traditional Chinese Medicine, The Fourth Clinical College of Xinjiang Medical University, Urumqi 830000, China.
Zhanjun Shu
Infectious Diseases and Nephropathy, The Fourth Clinical College of Xinjiang Medical University, Urumqi 830000, China.
Email: shu5857872@163.com
Received : March 27, 2023,
Accepted : May 04, 2023
Published : May 08, 2023,
Archived : www.jclinmedcasereports.com
Abstract
Since the spread of coronavirus disease 2019 (COVID-19) around the world, more than 630 million people have been infected and more than 6 million have died, posing a huge threat to human life and health. COVID-19 not only damages the respiratory system, but also affects other organs, including the kidney. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) accumulates in the kidney and causes damage through a variety of mechanisms. About 60% of the patients had different degrees of renal function changes and abnormal urine analysis. 5% to 80% of patients can develop Acute Kidney Injury (AKI), such as acute tubular necrosis and collapsing glomerulopathy, which increases the risk of death. COVID-23 19 patients with kidney injury will also indirectly increase the risk of serious complications related to COVID-19, including other symptoms such as arrhythmia, shock, and acute respiratory distress syndrome (ARDS). etc. At present, the problem of concurrent kidney injury in COVID-19 patients has been widely discussed and concerned. Here, we will discuss the clinical manifestations and pathogenesis of COVID-19 patients with kidney injury to provide a reference for clinical decision-making.
Keywords: Kidney injury; COVID-19; SARS-CoV-2; Clinical manifestations; Pathogenesis.
Copy right Statement:Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Shu Z (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Na X, Lu Z, Yaku Z, Qian Q, Li Y, Liu X, Shu Z, et al. Kidney injury in COVID-19 patients: Clinical manifestations and pathogenesis. Open J Clin Med Case Rep. 2023; 2033.
Introduction
Since December 2019, a new viral infectious disease swept the globe, caused a large global outbreak and was a major public health issue. International Committee on Taxonomy of Viruses (ICTV) announced “severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)” as the name of the new virus on 11 February 2020. At the same time, theWHO announced “COVID-19”as a name for the new illness [1].
According to the recent study of SARS-CoV-2, this virus not only attacks lungs, but also causes kidney damage. Abnormal kidney function and urinalysis are clinically visible and typically progress to Acute Kidney Injury (AKI). Recent studies have suggested that kidney damage can cause multiple organ dysfunction syndrome (MODS) and is associated with increased morbidity and mortality [2,3]. There is also evidence that even mild impairment in kidney function is an independent risk factor for COVID-19 infection and death [4].
Therefore, it is necessary to explore the clinical features 45 and mechanisms of kidney injury in COVID-19. This enables early intervention and treatment to ensure quality of life for patients.
Clinical Manifestations
Epidemiology
Recently, studies have shown that AKI is the common among kidney injury in hospitalized COVID-19 patients. The incidence of AKI ranges from 0.5% to 80%.Variability in AKI occurrence has been attributed to differences in clinical context, parameters used to diagnose AKI, geographic locations, race/ethnicity and severity of the disease, etc [5,6].
Older age, male sex, obesity, cardiovascular disease, Chronic Kidney Disease (CKD), kidney transplant, ICU patients, and people with multiple underlying diseases are risk factors for the development of AKI [7-10]. COVID-19 patients with AKI have a higher risk of ICU admission and increased requirements for mechanical ventilation and Renal Replacement Therapy (RRT). These are independent risk indicators for death in AKI patients. Some patients may experience hospital-acquired complications that lead to MODS or death. The more serious of COVID-19, the higher occurrence of AKI. Even AKI patients who survive to discharge from the hospital are at risk for the development of CKD or End-Stage Kidney Disease (ESKD) [11].
In a retrospective study, Gutierrez investigated 7,037 COVID-19 patients. The incidence of AKI was 10.87% and the mortality rate of AKI patients was twice than that patients without AKI [12]. Rahimzadeh evaluated 194 67 AKI patients, 20.1% of whom were in stage 3 and showed a significantly higher mortality rate, ARDS, required RRT and need for invasive ventilation than other stages [9]. Similar results were more pronounced in critically ill patients. Anandh et al. suggested that 1,714 ICU patients, 393 (22.9%) had severe AKI (AKIN stage 3) and all patients required RRT. The mortality rate for these AKIN3 patients was 52.4% [13]. Schaubroeck analyzed 1,286 critically ill COVID-19 patients, 85.1% with AKI and 9.8% with CRRT. All AKI stages were associated with ICU mortality [10].
Subsequently, researchers focused on AKI patients’ clinical prognosis. Some studies found that only 28.2% of patients recovered from AKI, while 71.8% did not fully recover upon discharge, and a small number of patients were discharged with AKD [9,14]. Other studies suggested that patients in the ICU and AKIN stage 3 had longer recovery times, most of whom still required dialysis upon discharge, and a higher level incidence of CKD [11]. In addition to the above studies, Huang followed up discharged COVID-19 patients for six months and showed a decrease in eGFR during follow-up in 13% of hospitalized patients without AKI and with normal eGFR [15].
Overall, COVID-19 patients who develop AKI will lead to reduced patient survival opportunities and survival quality. Therefore, early diagnosis and prevention of renal damage progression is important, which could improve clinical prognosis, and reduce the clinical progression of CKD/ ESKD.
Laboratory Features
Laboratory features in COVID-19 kidney injury patients 89 were characterized by abnormal indices such as proteinuria, hematuria, serum creatinine (Scr) and blood urea nitrogen (BUN) levels, etc. According to existing research, approximately 60% of COVID-19 patients show symptoms of kidney injury, but most do not progress to AKI [8].
Cheng analyzed urine and renal function tests of 710 COVID-19 patients in Wuhan, which showed that 43.9% of patients had proteinuria and 26.7% had hematuria. The proportion of elevated Scr patients was 14.4%, elevated BUN patients was 13.1% and estimated glomerular filtration rate (eGFR) <60 ml/min/1.73 m2 was 13.1% [7]. Xu and his colleagues found that COVID-19 AKI patients had lower platelet counts, lymphocyte counts, albumin levels, and serum calcium levels compared to patients without AKI, but had higher levels of leukocyte counts, neutrophils, and serum potassium [16]. Current clinical studies had demonstrated that elevated Scr, BUN, eGFR <60 ml/min/1.73 m2, lymphocyte count <1.5x109/L, leukocyte count >10x109/ L, proteinuria, and hematuria were independent risk factors for kidney injury and death. Higher Scr and PCT levels could predict the risk of AKI in COVID-19 patients. In addition, patients who were admitted with abnormal kidney function had a higher risk of worsening the disease [7,16]. Except for kidney function, urine analysis abnormalities were associated with COVID-19. Urine abnormalities on admission may predicted the degree of disease deterioration [17]. Schnabel found that the urinary albumin-to-creatinine ratio and serum albumin can predict AKI in COVID-19 patients. The higher 111 proteinuria during follow-up may point toward tubular damage [18]. Thus, some researchers believe urine analysis could be a better way to predict patient outcomes than kidney function [10]. Therefore, it is important to focus on renal function and urinalysis during hospitalization. For discharged kidney injury patients, persistent follow-up is necessary and essential. This is especially true for the long-term detection of renal function and routine urine tests.
Physiopathology
Current autopsy studies indicated that acute tubular injury was the most common finding in the kidneys of COVID-19 AKI patients. The other common histological findings were focal acute tubular necrosis [19], collapsing glomerulopathy, endothelial injury or thrombotic microangiopathy [20].
Sharma analyzed kidney biopsies from COVID-19 patients, all biopsy samples showed varying degrees of acute tubular necrosis, and small samples showed thrombotic microangiopathy, pauci-immune crescentic glomerular nephritis, and segmental glomerulosclerosis with features of healed collapsing glomerulopathy [21]. Postmortem examinations of COVID-19 patients also revealed acute proximal tubular damage and glomerular fibrin thrombi with ischemic collapse [22].
Collapsing glomerulopathy has been reported in several patients with COVID-19. Although the exact pathophysiology of collapsing glomerulopathy remains unknown, it may share common mechanisms with HIV-associated nephropathy, with podocyte injury through disruption of autophagy and mitochondrial homeostasis [23]. Similarly, some studies have suggested other mechanisms of collapsing glomerulopathy in COVID-19 patients. It may follow a host immune response involving the activation of interferon and chemokines, rather than direct infection of glomerular cells [24]. At the same time, multiple studies identified SARS-CoV-2 particles in kidney tissue. Viral RNA and protein were detected in the kidney [19,25]. In addition, SARS-CoV-2 particles have been observed in urine samples [26]. A series of kidney histology studies from China showed that these viruses not only have direct cytotoxic effects on kidney histology, but also initiate CD68 + macrophages and complement C5b-9 deposition, mediating renal tubule pathogenesis [25].
From a pathophysiological perspective, the proposed mechanisms of kidney injury include direct viral infection and indirect factors. SARS-CoV-2 has direct viral tropism to podocytes and proximal tubular cells. It can lead to acute glomerulonephritis, acute tubular necrosis, protein leakage in Bowman’s capsule, collapsing glomerulopathy and mitochondrial impairment. Indirect factors may arise from Renin-Angiotensin-Aldosterone System (RAAS) imbalance, coagulation dysfunction, cytokine storms and other factors associated with kidney injury in COVID-19 patients.
Pathogenesis
SARS-COV-2 and ACE2
Several studies indicated the genetic sequence analysis, and SARS-CoV-2 is genetically identical to 79% of SARS-CoV [27]. They all share the same cellular entry mechanism. Infection of human cell by specific binding of the 155 S-protein to the cell surface receptor Angiotensin-converting enzyme 2 (ACE2). The new study showed that the S protein in SARS-CoV was highly conserved in SARS-CoV-2 and that SARS-CoV-2 had 10 to 20 times more affinity for ACE2 than SARS-CoV. So SARS-CoV-2 was more infectious [28].
Notably, the mode of SARS-CoV-2 entry into cells is thought to be the binding of the viral spike S1 protein to the surface of ACE2. Forming a complex and through endocytosis or membrane fusion brings viral RNA into the cytoplasm. Then the virus began to replicate and transmit [28]. In addition to ACE2 being a widely recognized binding target, the entry process also involves the transmembrane protease serines 2 (TMPRSS2) and a disintegrin and metalloproteinase 17 (ADAM17), which together mediate the binding of SARS-CoV-2 to ACE2 on target cells, promoting viral membrane fusion and eventual entry into cells [29,30].
However, TMPRSS2 is more often in competition with ADAM17 [30]. The joint action of TMPRSS2 and ADAM17 is required to mediate cell endocytosis, but only TMPRSS2 is required to mediate membrane fusion without ADAM17 activity [28,30]. The virus replication efficiency of membrane fusion is 100 times higher than endocytosis [31].
Some studies have suggested that membrane fusion was the major mode of virus entry into cells. Only TMPRSS2 mediated membrane fusion can enhance SARS-CoV-2 entry cell [30]. TMPRSS2 may release cleaved S fragment into the extracellular fluid and inhibited antibody-mediated neutralizationt. Promoting the spread and incidence of the virus [32]. Conversely, ADAM17 shed ACE2 fragments (circulating ACE2), which are believed to protect host cells from viral infection. There is evidence that TMPRSS2 expression inhibits the effect of ADAM17 on ACE2. However, it is unclear how TMPRSS2 transcends ADAM17 to cleave ACE2 during SARS-CoV-2 infection [30]. Recently, two research groups demonstrated that ACE2 and TMPRSS2 were necessary for successful SARS-CoV-2 infection [29,33]. Some studies have shown that as long as a cell expresses both ACE2 and TMPRSS2, it has a higher risk of infection with SARS-CoV-2[34] (Figure 1(A)).
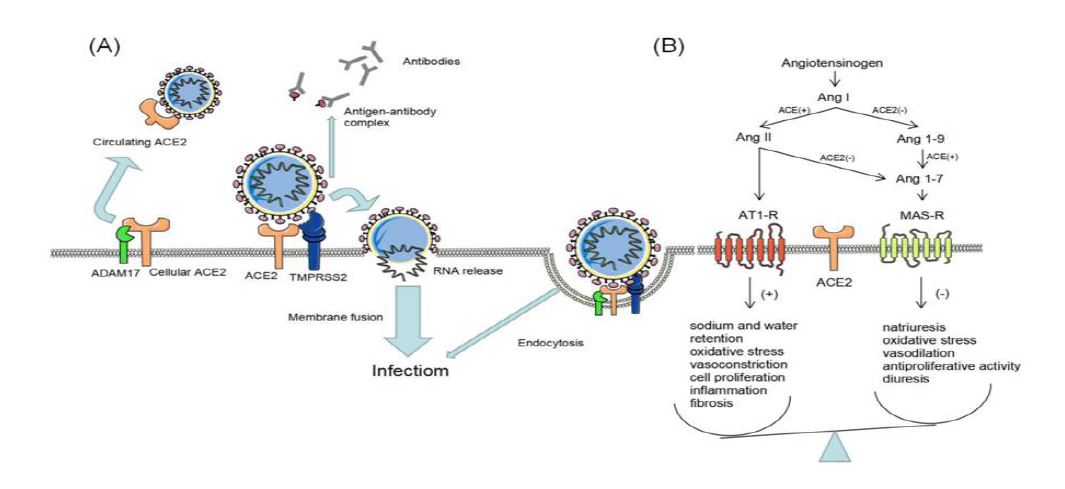
Figure 1: (A): SARS-CoV-2 can enter the host cells through 188 two possible pathways: membrane fusion and endocytic pathways. The membrane fusion is SARS-CoV-2 requires ACE2 and TMPRSS2 to cleavage the S protein. Then SARS-CoV-2 RNA is released into the cytoplasm and viral replication is efficiently processed. The endocytic pathways requires ACE2, TMPRSS2 and ADAM17 to support SARS-CoV-2 into cells. However, membrane fusion is the main mode of virus infection. ADAM17 participates in the production of circulating ACE2, releasing it into the extracellular fluid. Which protect host cell from the viral infection and counteract the effects of Ang II signaling. TMPRSS2 can cleaved S fragment and released into the extracellular fluid, then it binds to the antibody, inhibiting the antibody-mediated neutralization. To promote the spread and incidence of virus. (B): After SARS-CoV-2 entry into cells, the expression of ACE2 is significantly reduced and the basal level of ACE was upregulated. ACE2 / Ang- (1-7) / MasR protection mechanism is unbalanced, further leading to imbalance in the RAS system.
ACE2 and TMPRSS2 in Kidney
In the kidney, ACE2 is highly expressed in proximal renal tubules, podocytes and testicular stromal cells. ACE2 is weakly expressed in distal tubules and collection tubules [35]. TMPRSS2 has been detected to be highly expressed in the proximal tubules [36]. Pan clearly identified podocytes and proximal straight tubule cells as kidney host cells. SARS-CoV-2 enters host cells using ACE2 and TMPRSS, affecting the role of podocytes and proximal straight tubule cells in urine filtration, reabsorption and excretion [37].
There are also studies speculating that SARS-CoV-2 enters 210 host cells through CD147 mediated endocytosis. These findings suggest that CD147 and ACE2 may be complementary receptors that me diate viral infection [38]. CD147 is a ubiquitously expressed transmembrane glyco protein and is highly expressed in proximal tubular epithelial cells and inflammatory cells. It can be involved in renal tubule cell cycle regulation and the inflammatory response. So it can play an important role in different kidney diseases [39].
diate viral infection [38]. CD147 is a ubiquitously expressed transmembrane glyco protein and is highly expressed in proximal tubular epithelial cells and inflammatory cells. It can be involved in renal tubule cell cycle regulation and the inflammatory response. So it can play an important role in different kidney diseases [39].
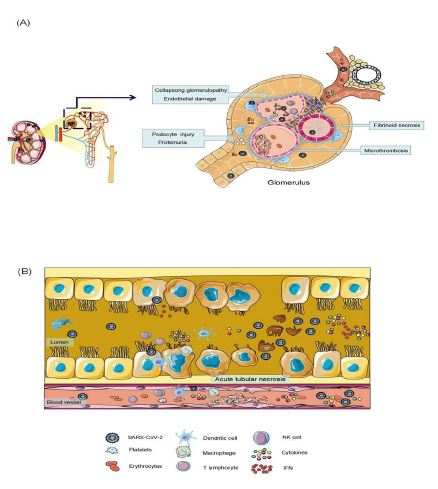
Figure 2: Glomerulus and renal tubules are the primary sites of SARS-COV-2 invasion. Proximal tubular epithelial cells cells and podocytes are the main host cells that SARS-CoV-2 directly invades. The virus enters the renal artery, first infecting glomerular endothelial cells, then podocytes. Podocyte dysfunction causes damage to an impaired glomerular filtration barrier, causing proteins and viruses to enter tubular fluid. Infection the renal proximal tubular epithelial cells leads to acute tubule injury. the SARS-CoV-2 causes glomerular collapse, glomerular fibrosis, acute tubular necrosis, endothelial injury, thrombotic microvascular disease.
Renin-Angiotensin-Aldosterone System (RASS) imbalances
In classical RASS, angiotensinogen is broken down by renin to give angiotensinⅠ (AngI), and angiotensin-converting enzyme (ACE) subsequently converts Ang Ⅰ to angiotensin Ⅱ (AngⅡ). In the kidneys, the ACE-AngⅡ-AT1 axis (the classical RAS) promotes sodium and water retention, oxidative stress, vasoconstriction, cell proliferation, inflammation, and fibrosis [41].
ACE2 is a key enzyme in RASS, which is involved in physiological regulation together with ACE. The main functions of ACE2 is to incorporate the inactive Ang1-9 from AngI and the catabolism of AngⅡ to form Ang1-7, which binds to the Mas receptor (R). Building the nonclassical RASS, the ACE2-Ang1-7-Mas axis, can counteract the effects of the ACE-AngⅡ-AT1 axis (the classical RAS) [41,42]. Specifically, it induces natriuresis, reduces oxidative stress, vasodilation, antiproliferative activity, and diuresis. These processes contribute to protecting the kidneys from damage [43].
However, after the binding of SARS-CoV-2 to the cell-surface receptor ACE2, ACE2 expression was significantly downregulated, and the basal level of ACE was upregulated. The degradation of AngII to Ang- 1-7 process is inhibited (less consumption), while AngI still continuously produces AngII (the generation is unchanged), leading to the accumulation of AngII in the body. Then the imbalance of the protective mechanism of ACE2 / Ang- (1-7) / MasR and further leads to the emergence of the RAS system imbalance. The clinical manifestations were excessive inflammatory response (including complement activation), vascular endothelial dysfunction, fibrosis status, and microthrombosis, etc. These factors can lead to kidney cell damage and increase the patient's chances of developing AKI [44] (Figure 1(B)).
Particularly in patients with CKD, Diabetic kidney disease (DKD) and ACE2 deficiency. The RASS is imbalanced, leading them to face a higher risk of AKI, acute tissue injury and even death.
Endothelial dysfunction and coagulation dysfunction
SARS-CoV-2 has been reported to specifically affect endothelial cells. Cases of COVID-19-related endodermatitis, microvascular or large vascular thrombosis have been reported several times. Renal endothelial cells can be directly infected with SARS-CoV-2, resulting in acute glomerular endothelial cell injury and thrombotic microvascular disease [45].
Laboratory indicators suggest that increased levels of plasma biomarkers of endothelial injury (e.g. soluble (s) E-selectin, sP-selectin, AngⅡ) and platelet activation (soluble thrombomodulin) are associated with poor prognosis [46]. Coagulation and fibrinolysis activation biomarkers (e.g. fibrinogen, PT, APTT, D-dimer) are associated with an increased risk of death in COVID-19 patients [13,14].
The mechanism of this phenomenon can be explored in the following way: (1) Immune-mediated, when the virus invades the body, the body develops an immune response and there are large amounts of virus particles and inflammatory factors in the blood. These stimulate and damage endothelial cells. Damaged endothelial cells raise more immune cells and activate the clotting pathway. The released chemokines and cytokines can induce capillary leak syndrome, microvascular inflammation and thrombosis leading to Disseminated Intravascular Coagulation (DIC). (2) RASS imbalances, after the SARS-CoV-2 infection, the RASS system is imbalanced. The local function of the AngII on the tissue will promote thrombosis, 289 inflammation, and tissue hypoxia. (3) Hypoxia, nitric oxide is a major vasodilator and antithrombotic factor. Most Covid-19 patients have hypoxia. Hypoxia can reduce nitric oxide synthase activity and nitric oxide bioavailability, cause constriction of blood vessels, abnormal perfusion and increased blood viscosity, which promotes thrombosis.
Therefore, endothelial impairment and coagulation dysfunction can increase the risk of micro‐infarctions in different organs, such as the heart, liver, and kidneys. Especially in patients with a history of cardiovascular disease patients, we should pay more attention to the coagulation function of patients.
Immune damage and a cytokine storms
During SARS-CoV-2 infection, viruses and drugs will cause the immune system to overactive. Laboratory examinations showed a decrease in the count of CD4+ and CD8+ T cells and a high secretion of chemokines and cytokines such as IL-6, IL-10, TNF-α, GM-CSF, IFN-γ, CCL-2, CCL-3, MCP1 and others. This can induce a severe systemic response, leading to cytokine storms and multiorgan damage [47].
Chemokines and cytokines play a key role in the kidney. They bind to receptors on the surface of kidney cells, triggering intracellular signaling pathways that lead to inflammatory cascades and cause renal endothelial dysfunction, microcirculatory dysfunction and renal tubule injury [48]. Gradin et al. examined the cytokine content in the urine of COVID-19 patients and found that 31 cytokines were associated with the stage of AKI and 19 were associated with maximal creatinine [49].
Beside, Cytokine storm can activate hemophagocytosis 310 occurs, which can observe the erythrophagocytosis and anaemia, inducing coagulation mechanism disorder, hypovolaemia or renal anemia, etc [2].
Decreased lymphocyte levels are common in COVID-19 patients, suggesting compromised immune function. At this point, the patient is susceptible to a variety of bacterial and fungal infections, which can lead to secondary infections and even progress to sepsis and burden the kidneys [50].
Both in the clinical and experimental models, scholars had observed the deposition of strong complement C5b-9 (membrane attack complex) in the renal tubules [50]. Jamaly et,al measured the high expression levels of deposited complement components(C1q,C3, FH,C5b-9), immunoglobulin, spleen tyrosine kinase (Syk), mucin-1 (MUC1) and calcium/calmodulin-dependent protein kinase IV(CaMK4) in the kidneys of COVID-19 patients[51]. These results suggest that activation of the renal complement pathway may exacerbate inflammatory responses and lead to kidney tissue injury.
Therefore, the current immune-mediated kidney injury mechanism as follows: (1) After SARS-CoV-2 infection of alveolar epithelial cells, resulting in an immune response and the associated chemokines and cytokines circulate into the bloodstream. A large number of inflammatory factors in plasma bind to receptors on the surface of kidney cells, disrupting signaling pathways within the cells. It triggers an inflammatory cascade that can damage kidney cells, leading to abnormal kidney function and endothelial dysfunction. (2) SARS-CoV-2 directly invades kidney target cells (proximal tubular cells, podiatric cells), leading to the 332 accumulation of large numbers of immune cells and cytokines in the kidney and the activation of the complement system and cytokine storm. Co-mediates kidney injury. (3) As the patient's illness time lengthens, the patient appears the immune exhaustion, which can lead to secondary infection, and even cause secondary kidney damage. At the same time, tissue hypoxia and renal anemia are also influential factors in kidney injury.
Other factors
Rhabdomyolysis: Rhabdomyolysis may be a risk factor for kidney damage in COVID-19 patients. When skeletal muscle is damaged, the breakdown product is released into the bloodstream, causing patients to develop AKI. In particular, some patients may have extensive subclinical renal impairment despite no significant AKI expression [22,52]. However, the exact mechanism of rhabdomyolysis in COVID-19 patients is unclear. At present, the different hypothesis is proposed for the mechanism of rhabdomyolysis induced by the virus: (1) direct invasion of skeletal muscle by the virus can lead to necrosis of muscle tissue. (2) Viral toxins in the bloodstream damage muscle cells. (3) Immune over activation caused by damaged muscle tissue can induce cytokine storms that can cause lasting damage to the body.
Hypoxia: Kidney is the second dirtiest oxygen drain after the heart. Therefore, the kidney is very sensitive to hypoxic injury [53]. The vast majority of critically ill COVID-19 patients may develop Acute Respiratory Distress Syndrome (ARDS), which causes the kidneys to become starved of oxygen. Acute hypoxemia can increase renal vascular resistance, lead to inadequate renal perfusion, abnormal kidney function and AKI [54].
In chronic hypoxia, endoplasmic reticulum and mitochondria develop oxidative stress that damages DNA, proteins and lipids in kidney cells. Causes inflammation, tissue fibrosis and mediates apoptosis of kidney cells, causing kidney damage, AKI and CKD progression [55]. AKI induced by hypoxia can lead to blood volume imbalance, electrolyte and acid-base levels disorders. Causes systemic hemodynamic changes and multi-organ dysfunction, forming a vicious circle. In addition, critically ill patients often require ventilator assisted ventilation when they are deprived of oxygen. Effects of mechanical ventilation on renal hemodynamics, nervous system, and immune-mediated processes may contribute to the development of AKI. The studies confirm that invasive ventilation is a risk factor for AKI [56].
Nephrotoxins drugs: Drug-induced kidney damage may also be an important factor in the development of COVID-19 patients. Several antiviral and antibiotic drugs have been widely used in the treatment of COVID-19, such as remdesivir, paxlovid, molnupiravir [57].
WHO revealed that the risk of AKI with remdesivir is 20 374 times higher than with other drugs commonly used for COVID-19 [58,59]. The use of some antibiotics has also been linked to an increased risk of AKI in COVID-19 patients [60]. Therefore, drugs with nephrotoxicity should be used with caution to reduce the burden on the kidneys and avoid kidney damage.
Conclusions
In summary, the pathogenesis of kidney injury in COVID-19 may be associated with SARS-CoV-2 target ACE2 mediated direct kidney injury, RASS imbalance, endothelial dysfunction, coagulation dysfunction, immune damage, and cytokine storm, el. However, most of the current research on COVID-19 with kidney injury has focused on clinical observation, and it is necessary to further explore the molecular mechanisms of kidney injury in animal models.
Second, COVID-19 patients should focus on early urine and kidney function testing. To determine if the patient has kidney damage, active measures can be taken to prevent the patient's condition from worsening multi-organ failure. Finally, for patients discharged after treatment, in addition to focusing on lung conditions, kidney function, kidney color ultrasound, and urine routine are also should be checked regularly. If a patient develops complications such as proteinuria, hematuria, and kidney function changes during follow-up, early and aggressive treatment should be used to delay the progression of kidney disease.
In the post-pandemic period, the continued mutation of SARS-CoV-2 has gradually made COVID-19 to a common infection. More and more scholars fully study it and the vaccine becomes widely available. We 396 should to keep the right attitude and get through this together.
Declarations
Conflicts of interest: All authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' contributions: Zhanjun Shu designed the study; Xie Na and Zhiwu Lu drafted and revised the manuscript; Zibiernisa Yaku and Qianqian Qian: performed the data curation and Investigation; Yue Li, Xiao Liu and SiqiWu: analyzed the data and make the visual pictures. All authors read and approved the final manuscript. Xie Na, Zhiwu Lu and Zhanjun Shu Contributed equally to this work.
Funding statement: This work was supported by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (grant numbers 2021D01A130);
Acknowledgments: The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
References
- Naming the coronavirus disease (COVID-19) and the virus that causes it. 2020.
- Diao B, Wang C, Wang R, Feng Z, Zhang J, et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. NAT COMMUN. 2021; 12: 2506.
- Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, et al. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med. 2020; 383: 590-592.
- Liakopoulos V, Roumeliotis S, Papachristou S, Papanas N. COVID-19 and the kidney: time to take a closer look. INT UROL NEPHROL. 2022; 54: 1053-1057.
- Chan L, Chaudhary K, Saha A, Chauhan K, Vaid A, et al. AKI in Hospitalized Patients with COVID-19. JAM SOC NEPHROL. 2021; 32: 151-160.
- Silver SA, Beaubien-Souligny W, Shah PS, Harel S, Blum D, et al. The Prevalence of Acute Kidney Injury in Patients Hospitalized with COVID-19 Infection: A Systematic Review and Meta-analysis. Kidney Med. 2021; 3: 83-98.
- Cheng Y, Luo R, Wang K, Zhang M, Wang Z, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. KIDNEY INT. 2021; 97: 829-838.
- Jafari-Oori M, Fiorentino M, Castellano G, Ebadi A, Rahimi-Bashar F, et al. Acute Kidney Injury and Covid-19: A Scoping Review and Meta-Analysis. ADV EXPMED BIOL. 2021; 1321: 309-324.
- Rahimzadeh H, Kazemian S, Rahbar M, Farrokhpour H, Montazeri M, et al. The Risk Factors and Clinical Outcomes Associated with Acute Kidney Injury in Patients with COVID-19: Data from a Large Cohort in Iran. Kidney Blood Press Res. 2021; 46: 620-628.
- Schaubroeck H, Vandenberghe W, Boer W, Boonen E, Dewulf B, et al. Acute kidney injury in critical COVID-19: a multicenter cohort analysis in seven large hospitals in Belgium. CRIT CARE. 2022; 26: 225.
- Ng JH, JS Hirsch, A Hazzan, R Wanchoo, HH Shah, et al. Outcomes Among Patients Hospitalized With COVID-19 and Acute Kidney Injury.AM J KIDNEY DIS. 2021; 77: 204-215.
- Gutierrez-Abejon E, Martin-Garcia D, Tamayo E, Alvarez FJ, Herrera-Gomez F. Clinical Profile, Pharmacological Treatment, and Predictors of Death Among Hospitalized COVID-19 Patients With Acute Kidney Injury: A Population-Based Registry Analysis. Front Med (Lausanne). 2021; 8: 657977.
- Anandh U, Noorin A, Kazmi S, Bannur S, Shah S, et al. Acute kidney injury in critically ill COVID-19 infected patients requiring dialysis: experience from India and Pakistan. BMC NEPHROL. 2022; 23: 308.
- Sarwal A, Gomez E, Perez-Gutierrez V, Carlos A, Afzal A, et al. Renal recovery after acute kidney injury in a minority population of hospitalized COVID-19 patients: A retrospective cohort study. Medicine (Baltimore). 2022; 101.
- Huang C, Huang L, Wang Y, Li X, Ren L, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. LANCET. 2021; 397: 220-232.
- Xu Z, Zhang Y, Zhang C, Xiong F, Zhang J, et al. Clinical Features and Outcomes of COVID-19 Patients with Acute Kidney Injury and Acute Kidney Injury 465 on Chronic Kidney Disease. AGING DIS. 2022; 13: 884-898.
- Gross O, Moerer O, Rauen T, Bockhaus J, Hoxha E, et al. Validation of a Prospective Urinalysis-Based Prediction Model for ICU Resources and Outcome of COVID-19 Disease: AMulticenter Cohort Study. J CLIN MED. 2021; 10.
- Schnabel K, N Garam, N Ledo, N Hajdu, A Koczy, et al. Urinary albumin-to-creatinine ratio and serum albumin are predictors of acute kidney injury in non-ventilated COVID-19 patients: a single-center prospective cohort study. INT UROL NEPHROL. 2022.
- Schurink B, E Roos, T Radonic, E Barbe, C Bouman, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020; 1: e290-e299.
- Legrand M, S Bell, L Forni, M Joannidis, JL Koyner, et al. Pathophysiology of COVID-19-associated acute kidney injury. NAT REV NEPHROL. 2021; 17: 751-764.
- Sharma P, NN Uppal, R Wanchoo, HH Shah, Y Yang, et al. COVID-19-Associated Kidney Injury: A Case Series of Kidney Biopsy Findings. J AM SOC NEPHROL. 2020; 31: 1948-1958.
- Fanelli V, M Fiorentino, V Cantaluppi, L Gesualdo, G Stallone, et al. Acute kidney injury in SARS-CoV-2 infected patients. CRIT CARE. 2020; 24: 155.
- Larsen CP, TD Bourne, JD Wilson, O Saqqa, MA Sharshir. Collapsing Glomerulopathy in a Patient With COVID-19. Kidney Int Rep. 2020; 5: 935-939.
- Ng JH, V Bijol, MA Sparks, ME Sise, H Izzedine, et al. Pathophysiology and Pathology of Acute Kidney Injury in Patients With COVID-19. Adv Chronic Kidney Dis. 2020; 27: 365-376.
- Su H, M Yang, C Wan, LX Yi, F Tang, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. KIDNEY INT. 2020; 98: 219-227.
- Guan WJ, NS Zhong. Clinical Characteristics of Covid-19 in China. Reply. N Engl J Med. 2020; 382: 1861-1862.
- Lu R, X Zhao, J Li, P Niu, B Yang, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. LANCET. 2020; 395: 565-574.
- Xiao L, H Sakagami, N Miwa. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 12. 2020.
- Hoffmann M, H Kleine-Weber, S Schroeder, N Kruger, T Herrler, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. CELL. 2020; 181: 271-280.
- Heurich A, H Hofmann-Winkler, S Gierer, T Liepold, O Jahn, et al. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J VIROL. 2014; 88: 1293-1307.
- Matsuyama S, M Ujike, S Morikawa, M Tashiro, F Taguchi. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci U S A. 2005; 102: 12543-12547.
- Glowacka I, S Bertram, MA Muller, P Allen, E Soilleux, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J VIROL. 2011; 85: 4122-4134.
- Matsuyama S, N Nao, K Shirato, M Kawase, S Saito, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A. 2020; 117: 7001-7003.
- Ziegler CGK, SJ Allon, SK Nyquist, IM Mbano, VN Miao, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. CELL. 2020; 181: 1016-1035.
- Fan C, W Lu, K Li, Y Ding, J Wang. ACE2 Expression in Kidney and Testis May Cause Kidney and Testis Infection in COVID-19 Patients. Front Med (Lausanne). 2020; 7: 563893.
- Ransick A, NO Lindstrom, J Liu, Q Zhu, JJ Guo, et al. Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. DEV CELL. 2019; 51: 399-413.
- Pan XW, D Xu, H Zhang, W Zhou, LH Wang, et al. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med. 2020; 46: 1114-1116.
- Wang K, W Chen, Z Zhang, Y Deng, JQ Lian, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020; 5: 283.
- Chiu PF, SL Su, CC Tsai, CL Wu, CL Kuo, et al. Cyclophilin A and CD147 associate with progression of diabetic nephropathy. Free Radic Res. 2018; 52: 1456-1463.
- Qi F, S Qian, S Zhang, Z Zhang. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. 2020; 526: 135-140.
- Mizuiri S, Y Ohashi. ACE and ACE2 in kidney disease. World J Nephrol. 2015; 4: 74-82.
- Patel VB, JC Zhong, MB Grant, GY Oudit. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. CIRC RES. 2016; 118: 1313-1326.
- Burns KD. The emerging role of angiotensin-converting enzyme-2 in the kidney. Curr Opin Nephrol Hypertens. 2007; 16: 116-121.
- Malha L, FB Mueller, MS Pecker, SJ Mann, P August, et al. COVID-19 and the Renin-Angiotensin System. Kidney Int Rep. 2020; 5: 563-565.
- Fox SE, FS Lameira, EB Rinker, HR Vander. Cardiac Endotheliitis and Multisystem Inflammatory Syndrome After COVID-19. ANN INTERN MED. 2020; 173: 1025-1027.
- Goshua G, AB Pine, ML Meizlish, CH Chang, H Zhang, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. LANCET HAEMATOL. 2020; 7: e575-e582.
- Wan S, Q Yi, S Fan, J Lv, X Zhang, et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br J Haematol. 2020; 189: 428-437.
- Mangalmurti N, CA Hunter. Cytokine Storms: Understanding COVID-19. IMMUNITY. 2020; 53: 19-25.
- Gradin A, H Andersson, T Luther, SB Anderberg, S Rubertsson, et al. Urinary cytokines correlate with acute kidney injury in critically ill COVID-19 patients. CYTOKINE. 2021; 146: 155589.
- Cybulsky AV, T Takano, J Papillon, A Khadir, J Liu, et al. Complement C5b-9 membrane attack complex increases expression of endoplasmic reticulum stress proteins in glomerular epithelial cells. J BIOL CHEM. 2002; 277: 41342-41351.
- Jamaly S, MG Tsokos, R Bhargava, OR Brook, JL Hecht, et al. Complement activation and increased expression of Syk, mucin-1 and CaMK4 in kidneys of patients with COVID-19. CLIN IMMUNOL. 2021; 229: 108795.
- Jin M, Q Tong. Rhabdomyolysis as Potential Late Complication Associated with COVID-19. EMERG INFECT DIS. 2020; 26: 1618-1620.
- Darmon M, F Schortgen, F Vargas, A Liazydi, B Schlemmer, et al. Diagnostic accuracy of Doppler renal resistive index for reversibility of acute kidney injury in critically ill patients. Intensive Care Med. 2011; 37: 68-76.
- Fogagnolo A, S Grasso, M Dres, L Gesualdo, F Murgolo, et al. Focus on renal blood flow in mechanically ventilated patients with SARS-CoV-2: a prospective pilot study. J CLIN MONIT COMPUT. 2022; 36: 161-167.
- Barthelemy R, V Beaucote, R Bordier, M Collet, A Le Gall, et al. Haemodynamic impact of positive end-expiratory pressure in SARS-CoV-2 acute respiratory distress syndrome: oxygenation versus oxygen delivery. Br J Anaesth. 2021; 126: e70-e72.
- Faour WH, A Choaib, E Issa, FE Choueiry, K Shbaklo, et al. Mechanisms of COVID-19-induced kidney injury and current pharmacotherapies. INFLAMM RES. 2022; 71: 39-56.
- Haddad F DGKR. A Comprehensive Review on the Efficacy of Several Pharmacologic Agents for the Treatment of COVID-19. Life (Basel). 2022.
- Gerard AO, A Laurain, A Fresse, N Parassol, M Muzzone, et al. Remdesivir and Acute Renal Failure: A Potential Safety Signal From Disproportionality Analysis of the WHO Safety Database. CLIN PHARMACOL THER. 2021; 109: 1021-1024.
- Binois Y, H Hachad, JE Salem, J Charpentier, B Lebrun-Vignes, et al. Acute Kidney Injury Associated With Lopinavir/Ritonavir Combined Therapy in Patients With COVID-19. Kidney Int Rep. 2020; 5: 1787-1790.
- Sang L, S Chen, X Zheng, W Guan, Z Zhang, et al. The incidence, risk factors and prognosis of acute kidney injury in severe and critically ill patients with COVID-19 in mainland China: a retrospective study. BMC PULM MED. 2020; 20: 290.
