
Open Access, Volume 9
Roles of BSEP protein in estrogen-induced intrahepatic cholestasis of pregnancy
E Liao1,2; Xiaomei Huang1; Siyu Chen1; Yong Shao1*
1The Department of Obstetrics and Gynecology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
2School of Manufacturing Science and Engineering, Southwest University of Science and Technology, Mianyang City, Sichuan Province 621010, People’s Republic of China.
Yong Shao
The Department of Obstetrics and Gynecology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Tel: 13101280793; Email: cqshaoyong@163.com
Received : January 18, 2023,
Accepted : February 15, 2023
Published : February 20, 2023,
Archived : www.jclinmedcasereports.com
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Shao Y (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Liao E, Huang X, Chen S, Shao Y. Roles of BSEP protein in estrogen-induced intrahepatic cholestasis of pregnancy. Open J Clin Med Case Rep. 2023; 1982.
Introduction
Intrahepatic Cholestasis of Pregnancy (ICP) is a liver disorder characterized by maternal pruritus, raised serum bile acids, elevated liver enzymes and adverse fetal outcomes, mostly occurs at the end of the second trimester or in the third trimester of pregnancy. It is most common in South America, particularly in Chile, where reports the incidence of 10% early [1], and as high as 6% in Chongqing, Sichuan and the Yangtze valley [2]. ICP is characterized by its unpredictable and sudden intrauterine death, and its associated high cesarean section rate, high iatrogenic preterm birth rate, amniotic fluid fecal fetal distress, still-birth and other related risks are difficult problems for obstetrics at present [3].
Although pathogenesis has been extensively studied, it’s still unknown at present. The domestic and international research on pathogenesis mainly focuses on estrogen, genetics, environmental factors and immunity. There is an apparent key role thought to be played by reproductive hormones and abnormal bile acid homeostasis in the pathogenesis of ICP. For the following reasons, 1) The timing of ICP onset and bile acid elevation were consistent with the period of estrogen elevation [4], 2) The incidence of ICP in twin pregnancy is higher than that in single pregnancy [5], 3) Patients with non-gestational oral estrogen-induced cholestasis are more likely to develop ICP [6], 4) High doses of 17α-ethynylestradiol caused intrahepatic cholestasis in wild mice but not in ERα knockout animals [7]. This evidence suggests that high estrogen levels play a significant role in the pathogenesis of ICP. However, the specific mechanism of estrogen inducing ICP during pregnancy remains unclear.
Regulation of estrogen pathogenicity of ICP on gene transcription
At present, domestic and foreign studies on the pathogenesis of ICP induced by estrogen mainly focus on the regulatory mechanism at the gene transcription level, namely the signaling pathway of the ERα-FXR/RXR-bile acid synthesis gene/bile acid transporter gene (Figure 1) [8]. In a non-pregnancy state, bile acid can enter the liver nucleus and bind to FXR-RXR heterodimer, activating the promoter IR-1, promoting the expression of promoter AP-1, then inducing the transcription of ABCE11 and ABCB4 genes, and promoting the mRNA and protein synthesis of BSEP (Bile salt export pump, ABCB11) and MDR3 (ABCB4). Increased expression of bile acid transporter protein on the cell membrane surface promotes bile acid transport and excretion into the bile ducts. While during pregnancy, a high level of estrogen can enter into the liver nucleus to activate ERα, bind with FXR-RXR to inhibit FXR gene expression, and then inhibit SHP-1 expression, lower BSEP, NTCP, OATP1, OATP2, OATP4 mRNA and protein levels from gene transcription level [7], and promote the transcription of bile acid synthesis genes CYP7A1 and CYP7B1, resulting in bile acid deposition in liver cells [10].
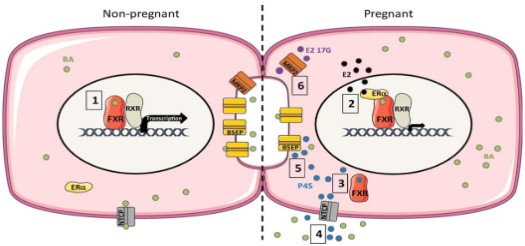
Figure 1: Estrogen’ effects on bile acid in hepatocytes [8]. In the non-pregnant state, bile acid binds to FXR and activates the
FXR-RXR complex, then promotes the transcription of the ABCB11 gene. During pregnancy, Estrogen binds to the ERα and
FXR-RXR complex, then inhibits the transcription of the ABCB11 gene.
BSEP is the main liver specific transporter in the hepatocytes to the bile ducts, followed by MRP2, MDR3, MDR1, AQP, etc. NTCP is the major liver specific transporter for the uptake of conjugated bile acids from portal blood, followed by OATPs and AQP. While OSTα-OSTβ is the main bile salt reabsorption receptor in the intestinal epithelial cells, followed by MRP3, MRP4 and OCT1 [11]. Among them, BSEP-mediated bile salt transport is the rate-limiting in the formation of bile salt dependent bile flow, and is the main driving force of enterohepatic circulation.
The role of BSEP in the pathogenesis of ICP
Defects in the quantity, structure and function of the ABCB11 gene can lead to the occurrence of hereditary or acquired cholestatic diseases, such as Progressive Familial Intrahepatic Cholestasis Type 2 (PFICII), Benign Recurrent Intrahepatic Cholestasis Type 2 (BRIC2), Drug-Induced Cholestasis (DIC). In recent years, gene sequencing of ICP patients has also found that about 20% of early-onset severe ICP have gene mutations such as ABCB11 (BSEP) or other bile acid transporters such as ABCB4 (MDR3) [12].
Structure and function of BSEP
Bile Salt Export Pump (BSEP) is encoded by the ABCB11 gene located on chromosome 2q23-31, encodes 1321 amino acids with a molecular weight of 140-170 kDa, belonging to the ATP Binding Cassette (ABC) transporter superfamily member. BSEP is located at the plasma membrane of the bile canaliculi at the top of hepatocytes, exists within lipid rich microdomains in canalicular membrane microvilii and in submembrane vesicles where it recycles as part of the mechanism that regulates its expression and function, mediates the 80% influx of bile acids into the bile ducts through the intracanalicular membrane of hepatocytes. Most of the known substrates of BSEP are endogenous hydrophilic bile salts [13]. Functional BSEP protein comprises at least four domains: two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs) in one polypeptide chain. The two TMDs form channels across the tubular membrane and are responsible for substrate binding and translocation of bile salts. The NBDs form a complex nucleotide binding site (NBS), with an ɑ-helical subdomain and a RecA-like ATPase core subdomain, which bind and hydrolyze ATP, and thereby provide energy for substrate transport of bile salts from the cytoplasm of hepatocytes to the bile canaliculi [14].
In the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/), more than 300 different ABCB11 gene mutations are currently identified in patients with cholestasis, including missense, nonsense, splicing abnormalities, deletions and insertions. Among them, BSEP shares about 48% structural identity with the other two structure-known transporters, ABCB1 (MDR1) and ABCB4 (MDR3), and these two transporters share about 71% sequence identity, but these three transporters differ significantly in substrate specificity [15]. MDR1 and MDR3 can respectively transport various organic cations and phosphatidylcholine, whereas BSEP selectively transports bile salts. However, the molecular mechanisms of substrate specificity on these three transporters are still unclear.
Liang Wang et al. [14] used Cryogenic Electron Microscopy (Cryo-EM) to observe the microscopic structure of BSEP (Figure 2), and linked 132 missense/nonsense single mutations to 300 reported clinical mutations. Among these 132 mutations, 97 missense mutations and 18 nonsense mutations are associated with the severe disease PFIC2, usually representing a complete loss of BSEP function. They also found 17 missense mutations cause the benign disease BRIC2, usually indicating partial retention function of BSEP.

Figure 2: a) The structure of ABCB11 is shown in two perpendicular views with cartoon representation. The N-terminal
helix’ electron density detail is shown in the rectangular box. b) A topology illustration of ABCB11[14].
Bile acid transporter gene mutations related to ICP
Jeremy S [16] summarized projects since 2004 on bile acid transporter gene sequencing in the ICP patients, covering 491 patients (261 controls) included in 2009 and 563 patients included in 2014 (642 controls) by Dixon, 310 patients with early-onset severe ICP enrolled by Turro in 2020. These projects showed that ICP was associated with ABCB11 (BSEP) and ABCB4 (MDR3) mutations.
Dixon’s study [17] of 491 ICP patients showed that the p.Val444Ala mutation of the ABCB11 gene was the most common point mutation. Turro’s study [18] showed that 20% of early-onset severe ICP were closely related to gene mutations. In summary, ABCB11 and ABCB4 gene mutations cause abnormal structure of BSEP and MDR3 proteins, leading to the inability to perform their functions and even turn them into components that need to be cleared by hepatocyte. Furthermore, Dixon et al. [19] summarized 6 most common mutations of ABCB11, involving rs2287622, rs2058996, rs7605199, rs3815676, rs3814382, rs7577650, and 6 most common mutations of ABCB4, involving rs2097937, rs31676, rs2097937, rs31676, rs1149222, rs4148826, rs2109505, rs2302386. Among them, rs3815676, rs7577650 and rs2109505 were selected as typical gene mutations.
Endoplasmic reticulum stress mechanism related to ICP
Some early-onset severe ICP are closely related to the faulty structure proteins produced by ABCB11 and ABCB4 mutation, while functional BSEP and MDR3 as seven-transmembrane proteins (7TMRs) on hepatocyte membranes, the synthesis, folding and membrane insertion are closely related to endoplasmic reticulum [13]. Newly synthesized BSEP undergoes N-linked core glycosylation in the endoplasmic reticulum, by mediating interactions with the lectin chaperones calnexin and calreticulin to increase the folding efficiency. Then, only correctly folded BSEP traffics to the Golgi apparatus in clathrin-coated COPII vesicles to fully glycosylated through trimmings to the core structure and extension from the core, to increase protein stability.
The Endoplasmic Reticulum-Associated Degradation (ERAD) mechanism recognizes aberrant folded proteins, induces ubiquitination and transports to the 26S proteasome for degradation [20]. Lin’ research [19] published on Hepatology in 2008 that focused on the 5 mutations of BSEP (G238V, D482G, G982R, R1153C, R1286Q) involved in FICII disease, and found that BSEP mutants (except R1153C) mainly degradation ERAD by the proteasome inordinately. All of these aberrant folded proteins present severe ubiquitination, manifested by elevated endoplasmic reticulum-localized Hed1, which targets the endoplasmic reticulum lumen E3 ubiquitin ligase of unfolded or misfolded proteins. Different designated E3 ligases are thought to recognize different BSEP mutants with substrate specificity [21]. In addition, Yong’s article [22] published on Trends Biochem Sci in 2017 elaborated on the recognition mechanism of unfolded or mis-folded proteins, which can be labeled by Lys48-linked polyubiquitin and mediated by designated E3 ligases for ubiquitination and further degradation by the proteasome. The ubiquitin-tagged substrates (mostly Lys48-linked and Lys63-linked) escaped from proteasomal degradation tend to form aggregates, which are specifically recognized and collected by specific adaptors of macroautophagy for lysosomal degradation.
At present, many studies have shown that the pathogenesis of ICP is closely related to the cell apoptosis mediated by Endoplasmic Reticulum Stress (ERS) [23]. Ting [24] conducted a whole proteomic analysis of 4 ICP patients and found that ERp29 was significantly increased in the placental, which is associated with the promotion of protein folding and endoplasmic reticulum transport. While the most important incentive of endoplasmic reticulum stress is unfolded or misfolded proteins.
The mechanism of PTM in the estrogeninduced ICP
Previous studies on the estrogen pathogenic theory of ICP mainly focused on the regulation of gene transcription: the promoter of ABCB11 gene is regulated by FXR, ERα, Nrf2 and other factors, which in turn affects BSEP’s transcription. However, more and more studies recently showed that transient regulation mechanism of bile acid transporters, namely the Post-Translational Modification (PTM), plays an important role in the early event of ICP [26]. In particular, estrogen [27], endotoxin (such as LPS) [28] and other stress conditions can induce abnormal localization of BSEP and MRP2 on the hepatocyte membrane. More precisely, hepatocyte membrane-resident BSEP and MRP2 experience the endocytosis through the Clathrin-Mediated Endocytosis (CME) under these stimulation, decreasing the proteins abundance on the cell surface by PTM, causing transporter disorders and intrahepatic cholestasis, which may be the initial cause of ICP.
The definite mechanism of early events in the estrogen pathogenic theory of ICP is not entirely understood. However, current research shows that estrogen induces ICP through regulation of gene transcription, considered to be a delayed response after abnormal bile acid transport, rather than an early event. Estrogen-induced endocytosis of hepatocyte membrane-resident BSEP may be the initial event of ICP.
Endocytosis mediated by estrogen through CME pathway
The estrogen receptors in human liver cells are mainly ERα and GPR30. Among them, GPR30 is located in the cell membrane, endoplasmic reticulum, Golgi apparatus and other membranous structures, and ERα is located in the nucleus. GPR30 (or GPER), a seven-transmembrane protein belonging to G Protein-Coupled Receptors (GPCRs), has a high affinity for estradiol, which is able to mediate rapid signal transduction within seconds to minutes independently of ERα, known as the rapid non-genomic effects of estrogen. The combination of estrogen and GPR30 causes the dissociation of G protein heterotrimer, which exerts effects such as cell proliferation and cell growth by activating the AC-cAMP-PKA pathway, Ca2+-dependent PKC pathway, releases HB-EGF and activates EGFR, and inhibits MAPKs p38α-ERK1/2 pathway, PI3K/PKB and other signaling pathways [29,30].
Endocytosis can be divided into phagocytosis, pinocytosis, and receptor-mediated endocytosis, according to the material size and the endocytosis mechanism. Among them, receptor-mediated endocytosis occurs in the form of clathrin-coated vesicles (CCV) [31]. The typical representatives are GPCRs that undergo signal attenuation and desensitization in this way. GPCRs-mediated endocytosis is minutely described in Alexander published in Nature Reviews Molecular Cell Biology in 2009 (Figure 3) [32]. Ligand-induced activation of GPCRs promotes signalling from the plasma membrane by phosphorylating the amino acid residue on the cytoplasmic side of the receptor, recruits β-statin and induces a conformational change of β-arrestin [33], and enables β-arrestin bind to the activated GPCRs as desensitized receptor–β-arrestin complexes to prevent further activation. The complexes are recruited to clathrin-coated pits (Clathrin-coated pits, CCP) by directly interacting with the clathrin coat adaptor complex AP2 or by binding to other adaptor proteins (eps15, eps15L1). Then, CCP invaginate inwards with the help of several accessory proteins and pinch off to form a Clathrin-Coated Vesicle (CCV) in a process that requires the GTPase dynamin. Internalized GPCRs face distinct trafficking destinations, recycle to the cell surface and re-associate with their G-proteins (resensitization) or target to ubiquitin-lysosomal system for degradation resulting in complete termination of receptor signaling (down-modulation). Filardo et al [34] also confirmed that GPR30-mediated endocytosis is consistent with GPCRs. Their study found that the GPR30 receptor was co-localized with clathrin after treating HEK-293 cells with 17β-estradiol, demonstrated that estradiol can activate GPR30 and receptor-mediated endocytosis through CME pathway.

Figure 3: Ligands activate GPCRs and induce receptor-mediated endocytosis mediated by the CME pathway [32].
Estrogen-induced endocytosis of BSEP on the hepatocyte membrane through the CME pathway
Gisel’s research [35] showed that estrogen can induce the internalization of BSEP through the CME pathway, reducing the abundance of BSEP and MRP2 on the hepatocyte membrane, resulting in bile acid transporter disorders and intrahepatic cholestasis. After they treated rat hepatocytes (IRHC) with estradiol-17β-D-glucuronide (E217G), the abundance of BSEP and MRP2 on the hepatocyte membrane decreased, which simulated the mechanism of pregnancy-induced cholestasis. Their further research [36] found that the three basic components of CME like AP2, clathrin, Rab5 co-localize with BSEP and MRP2, and the inhibition of AP2 expression by siRNA can completely prevent E217G-induced BSEP, MRP2 endocytosis. Andres’s study [37] clarified that estrogen induces the endocytosis of BSEP and MRP2 through the GPR30-AC-PKA pathway. After the GPR30 inhibitor G15 or RNAi technology was used to inhibit GPR30 expression, the abundance of BSEP and MRP2 on the cell membrane treated with E217G was consistent with the blank controller. Their study further found that inhibition of the GPR30-AC-cAMP-PKA pathway completely prevented E217G-induced internalization of BSEP and MRP2. And they first demonstrate that the activation of GPR30 is the initial event leading to the endocytosis of BSEP and MRP2.
Ortiz et al. [38] found that eps15 can promote the binding of BSEP to AP2, and experience endocytosed through the CME pathway. The membrane-localized BSEP increased by 94±17% after transfection of eps15 suppressor plasmid with MDCKII (Madin-Darby canine kidney) cells, but decreased by 21±12% after transfection of eps15 promoter plasmid. The BSEP mRNA levels both remained invariant. This suggests that the endocytosis of BSEP through the CME pathway requires the participation of eps15, which is one of the adaptor protein components in the CME pathway.
Ubiquitin modification of BSEP promotes endocytosis mediated by CME pathway
Current studies on the endocytosis mechanism of estrogen-induced BSEP through the CME pathway suggest that HAX-1[38], short-chain ubiquitination[39], C-terminal tyrosine sequence of bile acid transporters [40] and other factors that affect clathrin vesicle formation can affect the endocytosis process of BSEP. In fact, the endocytosed BSEP also faces distinct trafficking destinations as GPCRs, recycled to the hepatocyte membrane, or targeted to ubiquitin-lysosomal system for degradation. Estrogen can induce a persistent decrease in the abundance of BSEP on hepatocyte membrane. The endocytosed BSEP is likely to enter the protein degradation program. Therefore, ubiquitination of BSEP may play an important role in inducing CME pathway-mediated endocytosis.
Hisamitsu et al. [39] suggested that BSEP is modified by short-chain ubiquitination, which induced endocytosis of BSEP. Their study on the ABCB11 gene mutations of PFICII patients found that the two most common mutations (E297G and D482G) were highly ubiquitinated, resulting in a shortened half-life of BSEP on hepatocyte membrane. To identify this ubiquitination type, MDCK II cells were transfected with a recombinant adenovirus carrying the BSEP mutant cDNA, and induced BSEP to undergo ubiquitination modification. It was found that the original molecular weight of BSEP increased from 170 kDa to 190 kDa, while the molecular weight of a mature ubiquitin protein (Ub) is 8.5 kDa, suggesting that the shortened half-life of BSEP was modified by short-chain ubiquitination of 2-3 Ub molecules, resulting in membrane-localized BSEP. Further, Aida’s study [41] used UbΔDGG to replace the natural Ub, which lacks the C-terminal glycine of Ub, resulting in its inability to bind to substrate proteins and inhibit ubiquitination. Their study found that the expression of UbΔDGG prolongs the half-life of BSEP and MRP2 on the cell membrane, suggesting that the ubiquitination of BSEP and MRP2 induces the CME pathway mediated endocytosis. Studies [42] have shown that rifampicin-induced ubiquitination and proteasomal degradation of MRP2 mainly depend on E3 ligase GP78, and siRNA silencing GP78 can significantly inhibit rifampicin-induced MRP2 downregulation.
Conclusions
As for the pathogenesis of the ICP, the ABCB11 gene, which is closely related to bile acid transport, has attracted much attention. Based on gene transcription, the mutations of ABCB11 and endoplasmic reticulum stress induced may play an important role in the pathogenesis of ICP. At the same time, with the in-depth study of estrogen-induced intrahepatic cholestasis, more and more evidence indicatess that estrogen may induce the endocytosis of BSEP through the CME pathway through GPR30, and then reduces the abundance of BSEP on the hepatocyte membrane, resulting in bile acid transport disorder and intrahepatic cholestasis.
Declarations
Acknowledgements: We would like to thank all the patients and their families for participation in this study and for allowing this publication. We are grateful for the support of all the funding supportors including National Natural Sciences Foundation of China (No: 81471473) and Chongqing Health Planning Commission project (No. 2019ZDXM055).
Authors’ contributions: The corresponding author developed the study concept and design, critically reviewed the data and decided the content of the manuscript. All other authors acquired and/or analyzed data, performed statistical analysis and/or provided technical or material support and/or wrote and/or critically reviewed the manuscript. All authors finally decided the content of the manuscript.
Fundings: This work is funded by National Natural Sciences Foundation of China (No: 81471473) and Chongqing Health Planning Commission project (No. 2019ZDXM055). These fundings supported the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials: All data generated or analysed during this study are included in this published article.
Ethics approval and consent to participate: This study was approved by the ethical committee of the First Affiliated Hospital of Chongqing Medical University (2021-1826).
Consent for publication: Not applicable.
Competing interests: The authors declare that they have no competing interests.
References
- Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009; 15: 2049-2066.
- Qi HB, Shao Y, Wu WX et al. Grading of intrahepatic cholestasis of pregnancy. Zhonghua Fu Chan Ke Za Zhi. 2004; 39: 14-17.
- Ovadia C, Seed PT, Sklavounos A, Geenes V, Ilio CD, et al. Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: results of aggregate and individual patient data meta-analyses. Lancet. 2019; 393: 899-909.
- Leslie KK, Reznikov L, Simon FR, Fennessey PV, Reyes H, et al. “Estrogens in intrahepatic cholestasis of pregnancy. Obstetrics & Gynecology. 2000; 95: 372-376.
- Gonzalez MC, Reyes H, Arrese M, Figueroa D, Lorca B, et al. Intrahepatic cholestasis of pregnancy in twin pregnancies. J Hepatol. 1989; 9: 84-90.
- Williamson C, Hems LM, Goulis DG, Walker I, Chambers J, et al. Clinical outcome in a series of cases of obstetric cholestasis identified via a patient support group. BJOG. 2004; 111: 676-681.
- Yamamoto Y, Moore R, Hess HA, Guo GL, Gonzalez FJ, et al. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J Biol Chem. 2006; 281: 16625-16631.
- Abu-Hayyeh S, Williamson C. Estradiol, farnesoid X receptor, and altered metabolism in pregnancy. Hepatology. 2014; 60: 1815-1817.
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ, et al. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001; 276: 28857-28865.
- Milona A, Owen BM, Cobbold JF, Willemsen ECL, Cox IJ, et al. Raised hepatic bile acid concentrations during pregnancy in mice are associated with reduced farnesoid X receptor function. Hepatology. 2010; 52: 1341-1349.
- Boyer JL, Soroka CJ. Bile formation and secretion: An update. J Hepatol. 2021; 75: 190-201.
- Turro E, Astle WJ, Megy K, Gräf S, Greene D, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020; 24: 1-9.
- Sohail MI, Dönmez-Cakil Y, Szöllősi D, Stockner T. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. Int J Mol Sci. 2021; 22: 784.
- Wang L, Hou WT, Chen L, Jiang YL, Xu D, et al. Cryo-EM structure of human bile salts exporter ABCB11. Cell Res. 2020; 30: 623-625.
- Soroka CJ, Pate MK, Boyer JL. Canalicular export pumps traffic with polymeric immunoglobulin A receptor on the same microtubule-associated vesicle in rat liver. J Biol Chem. 1999; 274: 26416-26424.
- Nayagam JS, Williamson C, Joshi D, Thompson RJ. Review article: liver disease in adults with variants in the cholestasis-related genes ABCB11, ABCB4 and ATP8B1. Aliment Pharmacol Ther. 2020; 52: 1628-1639.
- Dixon PH, van Mil SWC, Chambers J, Strautnieks S, Thompson RJ, et al. Contribution of variant alleles of ABCB11 to susceptibility to intrahepatic cholestasis of pregnancy. Gut. 2009; 58: 537-544.
- Turro E, Astle WJ, Megy K, Gräf S, Greene D, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020; 24: 1-9.
- Dixon PH, Wadsworth CA, Chambers J, Donnelly J, Cooley S, et al. A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am J Gastroenterol. 2014; 109: 76-84.
- Needham PG, Guerriero CJ, Brodsky JL. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb Perspect Biol. 2019; 11: a033928.
- Wang L, Dong H, Soroka CJ, et al. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology. 2008;48(5):1558-1569.
- Kwon YT, Ciechanover A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem Sci. 2017; 42: 873-886.
- Xie RJ, Hu XX, Zheng L, et al. Calpain-2 activity promotes aberrant endoplasmic reticulum stress-related apoptosis in hepatocytes. World J Gastroenterol. 2020; 26: 1450-1462.
- Zhang T, Guo Y, Guo X, et al. Comparative proteomics analysis of placenta from pregnant women with intrahepatic cholestasis of pregnancy. PLoS One. 2013; 8: e83281.
- Brecker M, Khakhina S, Schubert TJ, et al.The Probable, Possible, and Novel Functions of ERp29. Front Physiol. 2020; 11: 574339.
- Kubitz R, Helmer A, Häussinger D. Biliary transport systems: short-term regulation. Methods Enzymol. 2005; 400: 542-557.
- Mottino AD, Crocenzi FA, Pozzi EJ. Role of microtubules in estradiol-17beta-D-glucuronide-induced alteration of canalicular Mrp2 localization and activity. Am J Physiol Gastrointest Liver Physiol. 2005; 288: G327-G336.
- Elferink MG, Olinga P, Draaisma AL. LPS-induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am J Physiol Gastrointest Liver Physiol. 2004; 287: G1008-G1016.
- Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007; 8: 462-470.
- Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol. 2014; 10: 1061-1065.
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009; 78: 857-902.
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009; 10: 609-622.
- Marchese A, Trejo J. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal. 2013; 25: 707-716.
- Filardo E, Quinn J, Pang Y, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007; 148: 3236-3245.
- Miszczuk GS, Barosso IR, Larocca MC. Mechanisms of canalicular transporter endocytosis in the cholestatic rat liver. Biochim Biophys Acta Mol Basis Dis. 2018; 1864: 1072-1085.
- Hayashi H, Inamura K, Aida K, et al. AP2 adaptor complex mediates bile salt export pump internalization and modulates its hepatocanalicular expression and transport function. Hepatology. 2012; 55: 1889-1900.
- Zucchetti AE, Barosso IR, Boaglio AC, et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ß-D-glucuronide-induced cholestasis. Hepatology. 2014; 59: 1016-1029.
- Ortiz DF, Moseley J, Calderon G, et al. Identification of HAX-1 as a protein that binds bile salt export protein and regulates its abundance in the apical membrane of Madin-Darby canine kidney cells. J Biol Chem. 2004; 279: 32761-32770.
- Hayashi H, Sugiyama Y. Short-chain ubiquitination is associated with the degradation rate of a cell-surface-resident bile salt export pump (BSEP/ABCB11). Mol Pharmacol. 2009; 75: 143-150.
- Lam P, Xu S, Soroka CJ, Boyer JL. A C-terminal tyrosine-based motif in the bile salt export pump directs clathrin-dependent endocytosis. Hepatology. 2012; 55: 1901-1911.
- Aida K, Hayashi H, Inamura K, et al. Differential roles of ubiquitination in the degradation mechanism of cell surface-resident bile salt export pump and multidrug resistance-associated protein 2. Mol Pharmacol. 2014; 85: 482-491.
- Xu BY, Tang XD, Chen J, et al. Rifampicin induces clathrin-dependent endocytosis and ubiquitin-proteasome degradation of MRP2 via oxidative stress-activated PKC-ERK/JNK/p38 and PI3K signaling pathways in HepG2 cells. Acta Pharmacol Sin. 2020; 41: 56-64.
