
Open Access, Volume 9
Jeune syndrome: Case report
Karina Elyane
Tecnológico de Monterrey, Mexico.
Email: karinaelyane07@gmail.com
Received : January 02, 2023
Accepted : February 01, 2023
Published : February 10, 2023,
Archived : www.jclinmedcasereports.com
Copy right Statement: Content published in the journal follows Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). © Elyane K (2023)
Journal: Open Journal of Clinical and Medical Case Reports is an international, open access, peer reviewed Journal mainly focused exclusively on the medical and clinical case reports.
Citation: Elyane K. Jeune syndrome: Case Report. Open J Clin Med Case Rep. 2023; 1978.
Introduction
Jeune syndrome, also known as asphyxiating thoracic dystrophy, was first described by Jeune et al in a couple of siblings back in 1955. He described 2 boys with certain distinctive characteristics, particularly a severely narrow thorax [1]. We now know it is a rare genetic condition presented in only 1 per 1000 000-130 000 live births in the USA [1]. Because of the lack of reported data, actual incidence in our country is not available in the literature.
It has been related to a mutation of different genes that cause different autosomal recessive skeletal ciliopathies including Jeune syndrome. Identified related genes with this condition are IFT80, TTC21B, WDR19 and DYNC2CH1 as the most frequent one. KIAA0753 mutations were recently described in children who fulfilled criteria for Jeune syndrome as well [2].
The diagnosis is made by clinical features and radiological findings including small narrow and rigid bell-shaped thorax, horizontal ribs, short tubular bones, micromelia, polydactyly, underdeveloped iliac wing (trident acetabulum), relatively large head, renal anomalies, and retinal degeneration [1,3]. Severity and clinical manifestations vary on each person due to genetic heterogeneity [3].
Survival rate after the neonatal period is poor [1]. It has been reported up to 90% of mortality in the neonatal period [3]. The most common cause of disease is respiratory failure because of alveolar hypoventilation due a restrictive thoracic cage [1]. The ones who survive the neonatal period may present in later stages of life renal, hepatic, ocular, and pancreatic complications [3].
We describe a case diagnosed by clinical and radiological features in a postnatal age.
Case Report
We present the case of a preterm male newborn of 35.5 weeks of gestation, born to a healthy 16 year old mother, non-consanguineous parents. The pregnancy was uneventful and there was no relevant family history with no history of genetic disorders. The prenatal care consisted of five visits and two ultrasounds, in which asymmetrical pelvic limbs in length were reported in the third trimester.
The baby was born by cesarean section due to a face presentation. At birth, the newborn had irregular respiratory effort, heart rate less than 100 beats per minute, acrocyanotic coloration and decreased muscle tone. The neonate required initial steps of neonatal resuscitation and five cycles of positive pressure ventilation. An improvement in heart rate, coloration and respiratory effort was obtained after the initial steps and the ventilation. The birth weight of the baby was 2.790 kg (62th centile Fenton), length being 46 cm, head circumference being 34 cm, chest diameter being 26 cm and abdominal perimeter being 29 cm.
After the birth, the infant was admitted to the Neonatal Intensive Care Unit due to respiratory distress and low oxygen saturation. Physical examination revealed hypertelorism, narrow chest, inverted nipples, and rhizomelic shortening in the upper limbs (Figure 1). The X-Ray of the chest revealed typical findings of narrow thoracic cage, short horizontal ribs, small iliac wings, and acetabulum with spurs (Figure 2). The Echocardiogram was normal. An assessment is made by the geneticist and suspicion of Jeune syndrome is established based on the findings on the physical examination and radiographs.
In the NICU, due to poor respiratory status, the neonate required mechanical ventilation with high parameters. The gasometries remained with abnormal results, maintaining oxygen saturation of 85-90%, paradoxical thoracic and diaphragmatic movements. The neonate continued the ventilatory support due to severe respiratory distress and died within 3 days as a result of progressive respiratory failure.
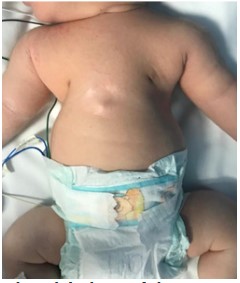
Figure 1: Clinical findings of the patient. Hypertelorism, narrow chest, inverted nipples, and rhizo-
melic shortening in the upper limbs.
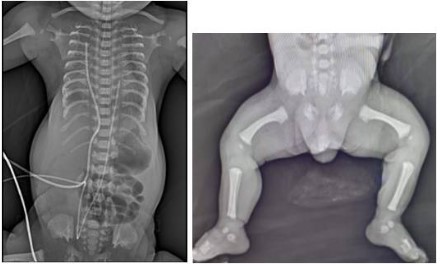
Figure 2: Anteroposterior (AP) plain films showing: (A) Narrow thoracic cage, short horizontal ribs, small iliac wings, and
acetabulum with spurs. (B) Trident acetabulum and short limbs.
Once removed, tissue from the left lower lobe nodule was tested for fungal cultures and with a gram stain. No fungus was isolated from the tissue culture at one and two weeks. The culture tissue with the gram stain showed no growth at four days, with few white blood cells and no observations of organisms.
Discussion
Jeune syndrome (also known as thoracic asphyxiant syndrome) is a rare skeletal ciliopathy in which there exist a primary dysfunction of the cilia [2,4]. It is known to be a hereditary condition with an autosomal recessive pattern [3]. Even Though the clinical presentation can vary between one individual and other, the classic clinical and radiological findings are: narrow thoracic cavity (bell shaped), short limbs, horizontal ribs, abnormal costochondral joints, trident appearance of the acetabula, elevated clavicle [3,4], some others may have kidney, liver or renal abnormalities and retinal degeneration [5].
The diagnosis is based on the clinical and radiological findings [4,6] and can be confirmed by genetic tests [4]. The most common gene mutation associated with this condition is DYNC2CH1 mutation, found in 50% of the cases [3]. In the present case we described a male baby who fulfilled clinical and radiographic criteria for Jeune syndrome. The patient’s clinical findings were hypertelorism, narrow chest, inverted nipples, and rhizomelic shortening of the upper limbs. The laboratory confirmation could not be done because of the precipitate demise of the patient and the lack of genetic tests in our hospital. Nevertheless, a medical assessment by a geneticist was made.
As part of the physical examination, we measured the Femur Length (FL), Thoracic Circumference (TC), Abdominal Circumference (AC), and TC/AC ratio in order to assess the spectrum of skeletal dysplasia. In this case we diagnosed micromelia (shortening of all tubular bones) [3]. The chest circumference resulted in 26 cm, that is <5th percentile the gestational age and the TC/AC was 0.89 cm. All this may indicate lung dysplasia [3].
Differential diagnosis with other skeletal dysplasia is easy, except for Ellis van-Creveld syndrome in which radiographic findings can be very similar. The difference found in Ellis Craveld syndrome is that the compromise of the thorax is not so noticeable [6]. In addition, Ellis van-Creveld syndrome is characterized by postaxial polydactyly (common), congenital heart defects and ectodermal dysplasia. Responsible mutations of the syndrome are in EVC and EVC2 genes [7].
Clinical spectrum is wide. In the severe form patients do not live beyond the neonatal period. It has been reported up to 80% of the patients die because of respiratory problems [6]. In the mild form, children develop complications in the consecutive years, mostly renal failure and tend to have recurrent respiratory infections, but it has been described that with time respiratory compromise decreases [6,8,9]. Inagas et al described a case of a 11 year old girl with diagnosis of Jeune syndrome who passed away due pneumonia associated with lung hypoplasia [4]. Our patient fits in the severe form.
In most cases the treatment consists of support measures such as endotracheal intubation, heart compressions, or oxygen supply [1]. Kids who survive to infancy may benefit from surgical treatment. Hoffman et al reported a total of 5 children with Jeune syndrome underwent surgical chest-expanding correction and concluded that indication for an operation is already given in newborns if failure of the right side of the heart and/or bronchopulmonary dysplasia do not allow fast improvement of their respiratory failure [10].
Conclusions
Jeune syndrome is a rare entity with an unfavorable prognosis. Prenatal diagnosis can be suspected by ultrasonic measurement. Diagnostic suspicion from the prenatal moment is essential for genetic counseling to the family. Jeune syndrome is sometimes compatible with life, although respiratory failure and infections are often fatal in infancy. Patients who survive infancy may benefit from multidisciplinary treatment including surgical treatment. It is important to increase the case report of this rare entity to pro- vide useful information and increase the therapeutic options that can be offered to those patients. It is also important to establish a correct diagnosis of this syndrome and then provide genetic counseling to parents due to the possibility of recurrent cases within the family.
References
- O’Connor MB, Gallagher DP, Mulloy E. Jeune syndrome. Postgraduate Medical Journal. 2008; 84: 559-559.
- Faudi E, Brischoux-Boucher E, Huber C, Dabudyk T, Lenoir M, et al. A new case of KIAA0753-related variant of Jeune asphyxiating thoracic dystrophy. European Journal of Medical Genetics. 2020.
- Phadke AK, Kumble A, Shafi S. Jeune syndrome: Asphyxiating thoracic dystrophy and its genetic diagnosis– A rare case report. Indian J Child Health. 2020; 7: 346-348.
- Holanda I, Ysa C, Olga N, Ruth S. Sindrome de Jeune: presentación de un caso clínico. Archivos Venezolanos de Puericultura y Pediatría. 2015; 78: 116-119.
- Phillips JD, van Aalst JA. Jeune’s syndrome (asphyxiating thoracic dystrophy): congenital and acquired. Seminars in Pediatric Surgery. 2008; 17: 167-172.
- De Vries J, Yntema JL, van Die CE, Crama N, Cornelissen EAM, et al. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. European Journal of Pediatrics. 2009; 169: 77-88.
- Ruiz-Perez VL, Stuart Tompson W, Helen Blair J, Espinoza-Valdez C, Lapunzina P, et al. Mutations in Two Nonhomologous Genes in a Head-to-Head Configuration Cause Ellis-van Creveld Syndrome. The American Journal of Human Genetics. 2003; 72: 728-732.
- CHANDAR V, ARORA A, NAGI H. ASPHYXIATING THORACIC DYSTROPHY. Medical Journal Armed Forces India. 1998; 54: 159-160.
- Naher BS, Saha BR, Naima KB. A Case Report on Jeune Syndrome in a Three-Month-Old Infant. Bangladesh Journal of Child Health. 2016; 39: 54-56.
- Hoffmann D, Hoffmann U, Hecker W. Zur Klinik und Problematik der Thoraxdystrophie. Klin.
